cdkbook
Stereochemistry
An intrinsic property of molecular structures is the 3D organization of the atoms,
resulting in a unique geometry. If we change the position of an atom, we get a
different geometry. We can lengthen and shorten a bond; change the angle
between two bonds; and rotate around bonds, changing torsion angles in
the molecules. All these changes lead to different conformations of the structure.
However, when we starting switching two atoms, or two atom groups, then we are no longer talking about conformations, but about stereoisomers. Stereoisomers share the same chemical graph, but no matter what combinations of bond and torsion changes, we cannot superimpose the two molecules on top of each other.
There are many kinds of geometrical constructs giving rise to stereochemistry. What they share, is that they are properties of the molecules, even though we are used to associate them with an atom or a bond. This is particularly the case for the two most common sources of stereochemistry: tetrahedral chirality, and double bond stereochemistry.
Stereochemistry in a flat world
Two dimensions drawings allow for depicting of three dimensional stereochemistry. It is not good at that, particular using wedge bonds, as us chemists typically do. In fact, there are so many ways it can go wrongs that a long list of guidelines have been developed [1].
The CDK has supported wedge bond stereo for a very long time, with its origin in the JChemPaint. An example 2D depiction is that of bromochlorofluoroiodomethane is shown in Figure 5.1.

Figure 5.1: 2D depictions can reflect stereochemistry using wedge bonds.
To add such 2D stereochemistry information we use the IBond.setStereo()
method:
Script 4.1 code/StereoisomerOne.groovy
isomer = new AtomContainer()
isomer.addAtom(new Atom("C"));
isomer.addAtom(new Atom("Cl"));
isomer.addAtom(new Atom("Br"));
isomer.addAtom(new Atom("F"));
isomer.addAtom(new Atom("I"));
isomer.addBond(0,1,Order.SINGLE)
isomer.addBond(0,2,Order.SINGLE)
isomer.getBond(1).setStereo(Stereo.UP)
isomer.addBond(0,3,Order.SINGLE)
isomer.getBond(2).setStereo(Stereo.UP)
isomer.addBond(0,4,Order.SINGLE)
This setStereo() and getStereo() methods in IBond take
and IBond.Stereo class. The values are defined by the matching enumeration
and can be iterated over and printed with:
Script 4.2 code/BondStereos.groovy
IBond.Stereo.each {
println it
}
That gives a fairly long list. Keep in mind that a bond is directed: a bond
has a first and second atom. That is the reason why an UP bond is
directed too. The thicker wedge side is at the side of the second atom.
If you like to invert that wedge bond, you use the UP_INVERTED
version.
But if we look at the full list, we also see that IBond.Stereo also
allows the specification of double bond stereochemistry:
NONE
UP
UP_INVERTED
DOWN
DOWN_INVERTED
UP_OR_DOWN
UP_OR_DOWN_INVERTED
E_OR_Z
E
Z
E_Z_BY_COORDINATES
However, nowadays the CDK also has other means to specify stereochemistry that is independent from 2D depictions, and those our outlined in the next sections.
Tetrahedral chirality
The tetrahedral chirality describes the geometry around four-coordinate atoms. For example, consider methane. It has five atoms, connected with four bonds. For the 3D geometry, a four-coordinate carbon gives a tetrahedral structure with the four atoms connected to the atoms at the four corners and a carbon right in the middle. Note that we can two switch hydrogen atoms, but that would not make any difference.
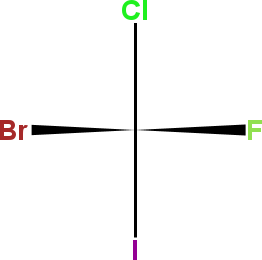
Figure 5.2: Stereoisomers of bromochlorofluoroiodomethane.
If we replace two hydrogens with a chloride and a bromide, we can still switch the two hydrogen atoms, and still have the same geometry. If we switch the two halogens from place, the structure depiction will change at first, but we would quickly notice that if we rotate one of the two structures, we still have the same structure.
The interesting turning point is when you replace the third hydrogen with yet another halogen. Now we have four different atoms surrounding the central atom. There is just a single chemical graph, with five atoms, connected with four bonds. For the 3D geometry, a four-coordinate carbon gives a tetrahedral structure with the four atoms connected to the atoms at the four corners and a carbon right in the middle.
With this geometry we no longer can switch two atoms bound to the carbon without changing the geometry: switching two halogens causes the stereochemistry to change. In fact, there are two possible stereoisomers, each of which is a mirror image of the other, as depicted in Figure 5.2.
Because wedge bonds are ambiguous and only work for systems with specified
2D coordinates, a data model has been set up that is independent from coordinate
systems. The base interface is IStereoElement from which specific
stereo elements derive. This is depicted in Figure 5.3.
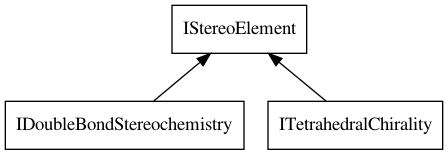
Figure 5.3: The ITetrahedralChirality and IDoubleBondStereochemistry interfaces extends the IStereoElement interface.
The ITetrahedralChirality interface requires you to specify the four neighboring
atoms around a central chiral atom. Thus for bromochlorofluoroiodomethane
we can define the chirality also without coordinates. For this, we use the
constructor of the interface’s prime implementation:
Script 4.4 code/TetrahedralStereo.groovy
isomer = new AtomContainer()
isomer.addAtom(new Atom("C"))
isomer.addAtom(new Atom("Cl"))
isomer.addAtom(new Atom("Br"))
isomer.addAtom(new Atom("F"))
isomer.addAtom(new Atom("I"))
ligands = new IAtom[4]
ligands[0] = isomer.getAtom(1)
ligands[1] = isomer.getAtom(2)
ligands[2] = isomer.getAtom(3)
ligands[3] = isomer.getAtom(4)
chirality = new TetrahedralChirality(
isomer.getAtom(0), ligands,
Stereo.CLOCKWISE
)
The interface provides appropriate getter methods for algorithms to use:
Script 4.5 code/TetrahedralIface.groovy
println "Central atom: " +
chirality.chiralAtom.symbol
println "Ligand atoms: " +
chirality.ligands[0].symbol + " " +
chirality.ligands[1].symbol + " " +
chirality.ligands[2].symbol + " " +
chirality.ligands[3].symbol
println "Stereo: " +
chirality.stereo
Which reports:
Central atom: C
Ligand atoms: Cl Br F I
Stereo: CLOCKWISE
Mind you, the Stereo class listed here is different from that for IBonds.
ITetrahedralChirality.Stereo has only two values, which we print like any
enumeration with:
Script 4.6 code/ChiralityStereos.groovy
ITetrahedralChirality.Stereo.each {
println it
}
Which shows that following seen from the first ligand atom, in order of the remaining three ligand atoms, they following a clockwise or anti-clockwise circle:
CLOCKWISE
ANTI_CLOCKWISE
References
- Brecher J. Graphical representation of stereochemical configuration (IUPAC Recommendations 2006). Pure and Applied Chemistry. 2006 Jan 1;78(10):1897–970. doi:10.1351/PAC200678101897 (Scholia)