metabolomics-practical
Pathway Analysis
| prev | toc | next |
Continuing from the data set we looked at in the previous section, we first have to define the two sitations we want to compare. Otherwise, we cannot saw which pathways are changed when comparing the two situations. Let’s compare the read blood cell samples with plasma:
rbcColumns <- c(
"Person4_RBC1_POS", "Person4_RBC2_POS",
"Person4_RBC3_POS", "Person4_RBC4_POS"
)
plasmaColumns <- c(
"Person4_Plasma1_POS", "Person4_Plasma2_POS",
"Person4_Plasma3_POS", "Person4_Plasma4_POS"
)
Using these two groups, we can calculate log fold changes (logFC) with:
logFC <- log2(
apply(mtbls88[,rbcColumns], 1, function(x) sum(x)) /
apply(mtbls88[,plasmaColumns], 1, function(x) sum(x))
)
hist(logFC, breaks = 50, col="gold")
The hist() command shows a histogram of of the fold changes:
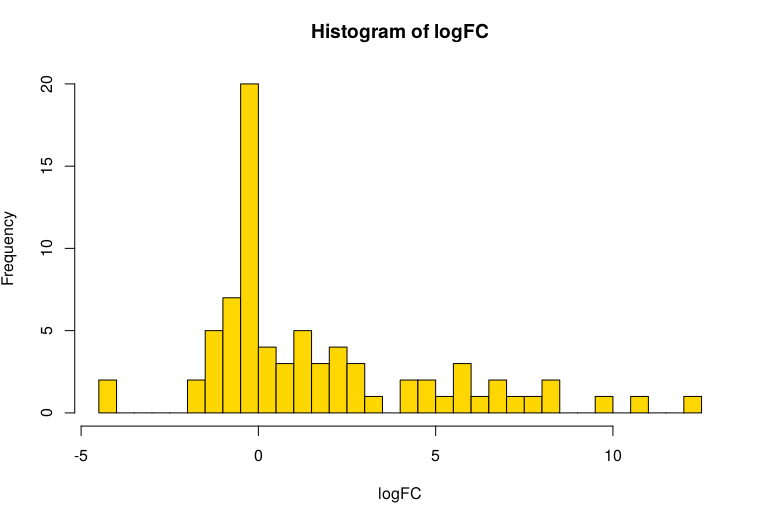
We are going to use this data in PathVisio (download). PathVisio needs Java 8 and both you may need to install first. Also, we need to export the data file as a TSV file first. We create a new data matrix, and leave out a few rows:
logFCdata <- cbind(
as.character(mtbls88[,"database_identifier"]),
logFC
)[-c(27,47,49,75,77),]
write.table(
logFCdata, file = "logfc.tsv",
sep = "\t",
col.names = c("ChEBI", "logFC"),
row.names = FALSE, quote = FALSE
)
Loading the fold changes into PathVisio
Following the same approach for pathway analysis with other omics data, we can use this data
to find potentially interesting pathways. First, download the
metabolite identifier database
and open that in PathVisio with Data, Select Metabolite Database.
Then, import the logfc.tsv file you created in the previous step into PathVisio
with Data, Import expression data. The file is TAB separated:
The first CHEBI column has the identifiers, and they are all ChEBI identifiers:
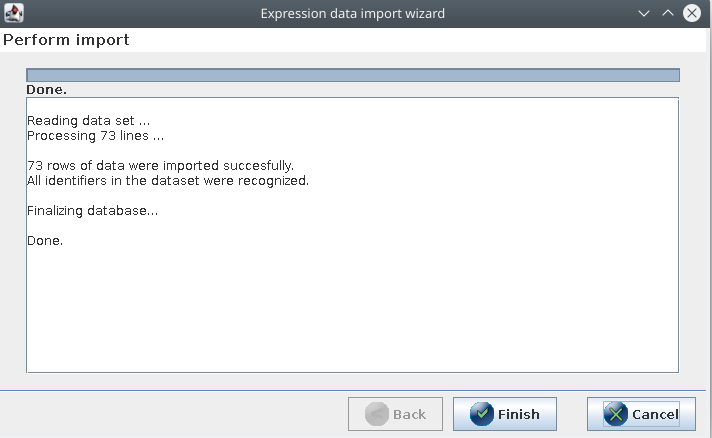
If all went well with loading the metabolite identifier database and importing the expression data, then all 73 data rows should be imported correctly and all identifiers recognized:
Finding pathways with PathVisio
The next step is to download the human pathways GPML archive and unzip the file locally. Maybe you have them still around from a previous practical. Then downloading them again is not needed.
The pathway analysis goes in exactly the same way as for genes, except that we do not
have p-values. The analysis with Data, Statistics... has a simpler equation. The list of
metabolites is similar (or use the equation [logFC] < -0.2 OR [logFC] > 0.2):
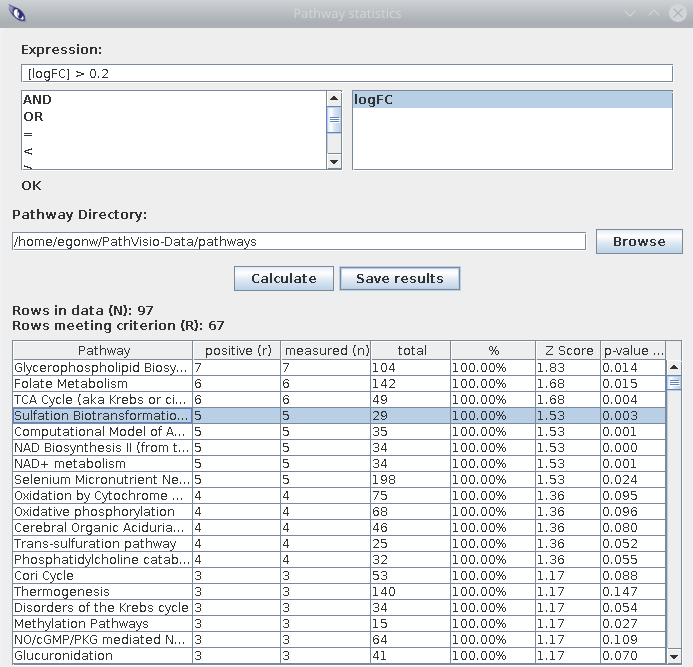
After calculating the enrichment, make sure pathways have non-zero values in the
positives and measured columnd. The second is the number of metabolites in this
pathway for which the experimental data has fold changes. The positives is
the number of measured metabolites that meet the expression.
Make sure to open the pathway to see what it looks like. For example, here is the experimental data mapped onto WP692:
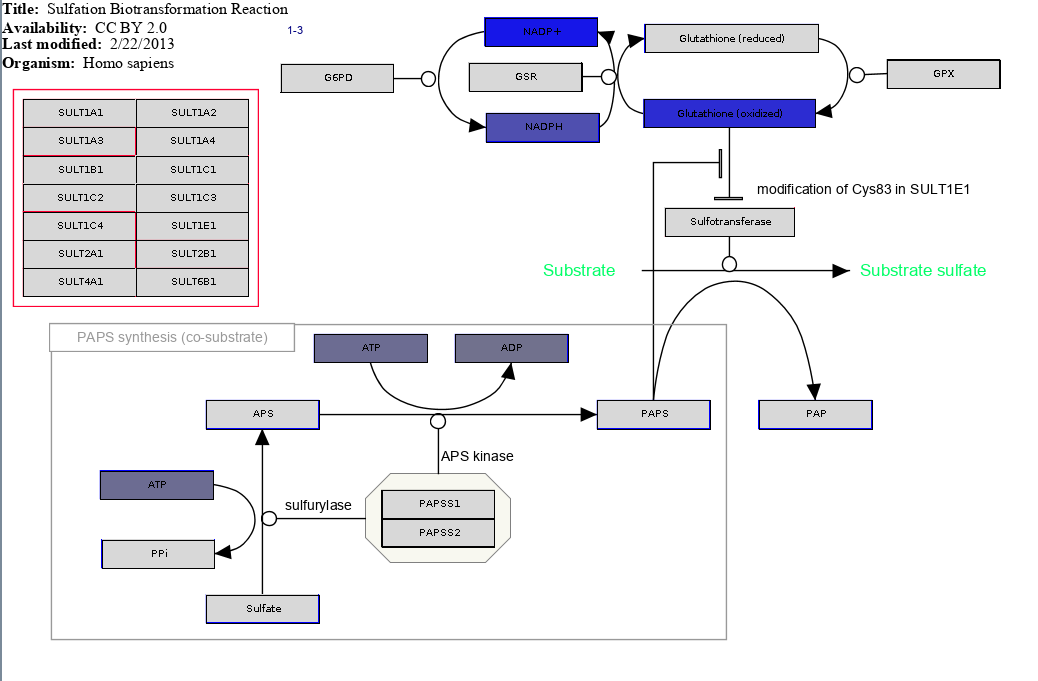
- would you expect to see experimental data for reduced glutathion? Maybe that could be expected. What are reasons why we do not have experimental data for it?
- if we look at the first few pathways, with the general patterns do we see? Metabolites like ATP/ADP and NADP/NADPH show up in many pathways. What is the biological meaning of this?
General thoughts
- are 73 metabolites enough to do pathway analysis? One problem is that many pathways have only few metabolites measured. These pathways may, however, allow you to search in the original experimental data for unidentified metabolites.
Open the TCA cycle pathway in PathVisio and click the NAD on the right side, and open the Backpage sidepanel in PathVisio, like this:
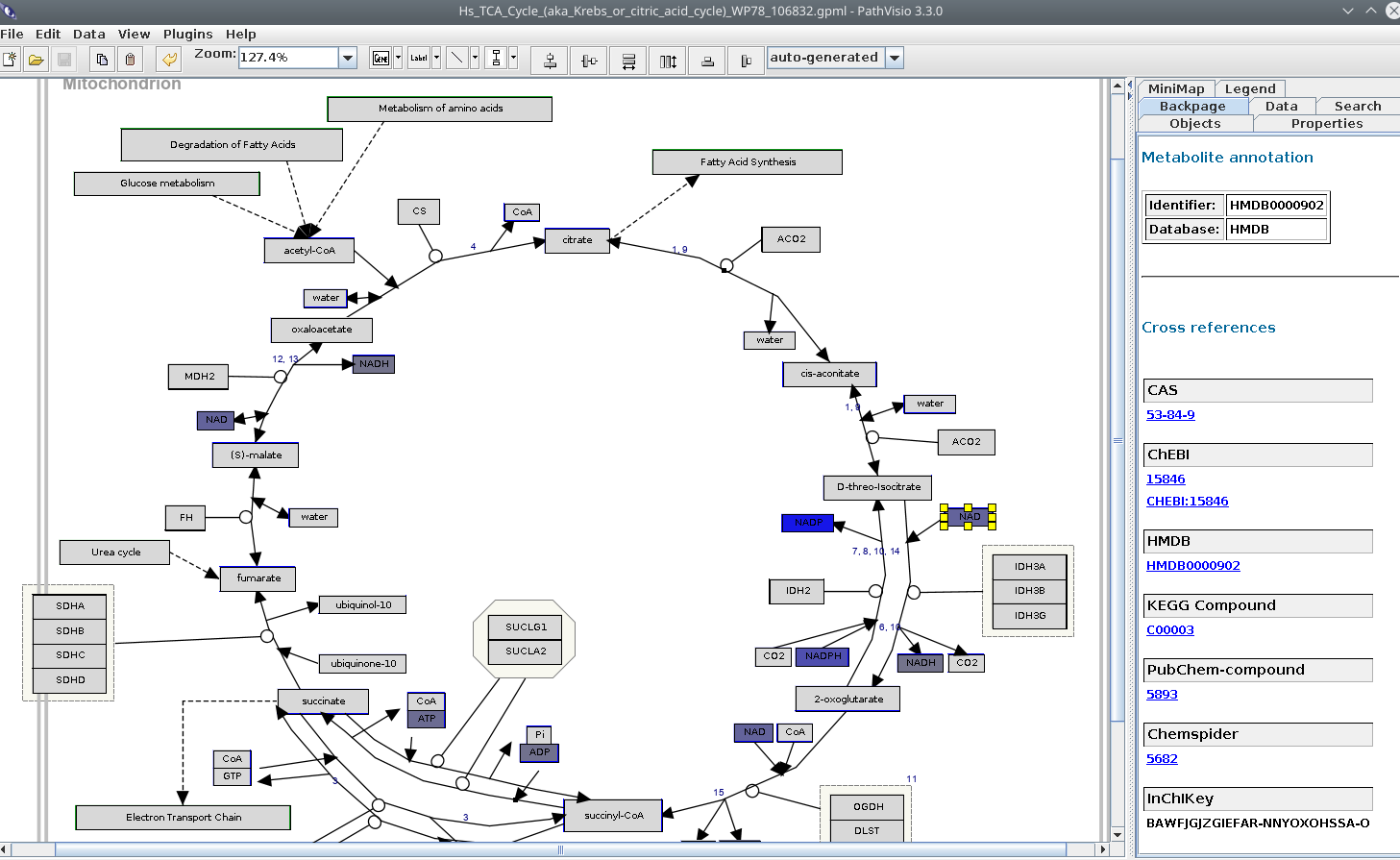
At the top we can see that the NAD node in the pathway is annotated with an HMDB identifier.
PathVisio used the BridgeDb metabolite identifier mapping database to recognize that the HMDB identifier in the pathway
and the ChEBI identifier for the experimental data actually are about the same metabolite. Then:
- thinking about the previous sections of this practical and the lecture, what other things could we do to improve this analysis? The current analysis is limited by the identifier mapping provided by the BridgeDb database, but this does not map structures with different stereochemistry or different charge states. This means that the mapping of experimental data onto the pathways is not always done when it actually should. For example, if the pathway drawing has a metabolite that does not specify the stereochemistry, or has the identifier of the charged metabolite, and the experimental data of the neutral metabolite.
| prev | toc | next |
Copyright 2020-2026 (C) Egon Willighagen Dept of Translational Genomics - CC-BY Int. 4.0