cdkbook
Atoms, Bonds and Molecules
The basic objects in the CDK are the IAtom, IBond and
IAtomContainer [1].
The name of the latter is somewhat misleading, as it contains
not just IAtoms but also IBonds. The primary use of the model is the
graph-based representation of molecules, where bonds are edges between
two atoms being the nodes [2].
Before we start, it is important to note that CDK 2.0 has an important
convention around object properties: when a property is unset, the
object’s field is set to null. This brings in sources for NullPointerExceptions,
but also allows us to distinguish between, for example, zero and unset
formal charge. In the former case, the formal charge value be set and have
a zero value; in the latter case, the field has a null value, indicating the
formal charge is currently unknown.
Atoms
The CDK interface IAtom is the underlying data model of atoms. Creating
a new atom is fairly easy. For example, we can create an atom of element
type carbon, as defined by the element’s atomic number that we pass as parameter
in the constructor:
Script 3.1 code/CreateAtom3.groovy
atom = new Atom(6);
For this we can also use the atomic number from the IElement class:
Script code/CreateAtom4.groovy
atom = new Atom(IElement.C);
An atom can also be constructed by passing in the symbol but this is marginally less efficient:
Script 3.2 code/CreateAtom1.groovy
IAtom atom = new Atom("C");
Alternatively, we can also construct a new carbon atom, by passing a
carbon IElement, conveniently provided by the Elements class:
Script 3.3 code/CreateAtom2.groovy
IAtom atom = new Atom(Elements.CARBON);
A CDK atom has many properties, many of them inherited from the IElement,
IIsotope and IAtomType interfaces. Figure 4.1 shows the interface
inheritance specified by the CDK data model.
These constructors will set the atomic number of the atom:
atomic number: 6
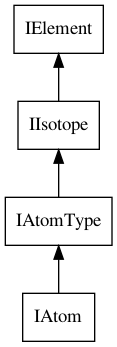
Figure 4.1: The IAtom interface extends the IAtomType interface, which extends the IIsotope interface, which, in turn, extends the IElement interface.
IElement
The most common property of IElements are their symbol and atomic
number. Because the IAtom extends the IElement, CDK atoms also have
these properties. Therefore, we can set these properties for atoms
manually too:
Script 3.4 code/ElementProperties.groovy
atom.setSymbol("N")
atom.setAtomicNumber(7)
Of course, we can use the matching get methods to recover the properties:
Script 3.5 code/ElementGetProperties.groovy
IAtom atom = new Atom(Elements.CARBON);
println "Symbol: " + atom.getSymbol()
println "Atomic number: " + atom.getAtomicNumber()
which outputs:
Symbol: C
Atomic number: 6
IIsotope
The IIsotope information consists of the mass number, exact mass and
natural abundance:
Script 3.6 code/IsotopeProperties.groovy
IAtom atom = new Atom("C");
atom.setMassNumber(13)
atom.setNaturalAbundance(1.07)
atom.setExactMass(13.00335484)
Here too, the complementary get methods are available:
Script 3.7 code/IsotopeGetProperties.groovy
println "Mass number: " + atom.getMassNumber()
println "Natural abundance: " + atom.getNaturalAbundance()
println "Exact mass: " + atom.getExactMass()
giving:
Mass number: 13
Natural abundance: 1.07
Exact mass: 13.00335484
Appendix B lists all isotopes defined in the CDK with a natural abundance of more than 0.1.
IAtomType
Atom types are an important concept in cheminformatics. They describe some basic facts about that particular atom in some particular configuration. These properties are used in many cheminformatics algorithms, including adding hydrogens to hydrogen-depleted chemical graphs (see Section 15.4.1) and force fields. Chapter 13 provides much more detail on the atom type infrastructure in the CDK library, and, for example, details how atom types can be perceived, and how atom type information is set for atoms.
The IAtomType interface contains fields that relate to atom types. These
properties include formal charge, neighbor count, maximum bond order
and atom type name:
Script 3.8 code/AtomTypeProperties.groovy
atom.setAtomTypeName("C.3")
atom.setFormalCharge(-1)
atom.setMaxBondOrder(IBond.Order.SINGLE)
atom.setFormalNeighbourCount(4)
Coordinates
The IAtom class supports three types of coordinates: 2D coordinates,
used for diagrams, 3D coordinates for geometries, and crystal unit cell
or notional coordinates. These properties are set with the respective
methods:
Script 3.9 code/AtomCoordinates.groovy
atom.setPoint2d(
new Point2d(1.0, 2.3)
)
atom.setPoint3d(
new Point3d(-2.0, -3.5, 4.7)
)
atom.setFractionalPoint3d(
new Point3d(0.1, 0.5, 0.25)
)
The latter coordinates define the locations of the atoms with respect to (or inside) the crystal structure’s unit cell. Section 5.2 explains the full crystal structure functionality.
Bonds
The IBond interface of the CDK is an interaction between two or more
IAtoms, extending the IElectronContainer interface. While the most
common application in the CDK originates from graph theory [2], it is not
restricted to that. That said, many algorithms implemented in the CDK
expect a graph theory based model, where each bond connects two, and
not more, atoms.
For example, to create ethanol we write:
Script 3.10 code/Ethanol.groovy
IAtom atom1 = new Atom("C")
IAtom atom2 = new Atom("C")
IAtom atom3 = new Atom("O")
IBond bond1 = new Bond(atom1, atom2, IBond.Order.SINGLE);
IBond bond2 = new Bond(atom2, atom3, IBond.Order.SINGLE);
The CDK has a few bond orders, which we can list with this groovy code:
Script 3.11 code/BondOrders.groovy
IBond.Order.each {
println it
}
which outputs:
SINGLE
DOUBLE
TRIPLE
QUADRUPLE
QUINTUPLE
SEXTUPLE
UNSET
As you might notice, there is no AROMATIC bond defined. This is
deliberate and the CDK allows to define single-double bond order patterns at
the same time as aromaticity information. For example, a kekule
structure of benzene with bonds marked as aromatic can be constructed with:
Script 3.12 code/AromaticBond.groovy
IAtom atom1 = new Atom("C")
IAtom atom2 = new Atom("C")
IAtom atom3 = new Atom("C")
IAtom atom4 = new Atom("C")
IAtom atom5 = new Atom("C")
IAtom atom6 = new Atom("C")
IBond bond1 = new Bond(atom1, atom2, IBond.Order.SINGLE)
IBond bond2 = new Bond(atom2, atom3, IBond.Order.DOUBLE)
IBond bond3 = new Bond(atom3, atom4, IBond.Order.SINGLE)
IBond bond4 = new Bond(atom4, atom5, IBond.Order.DOUBLE)
IBond bond5 = new Bond(atom5, atom6, IBond.Order.SINGLE)
IBond bond6 = new Bond(atom6, atom1, IBond.Order.DOUBLE)
bond1.setFlag(CDKConstants.ISAROMATIC, true);
bond2.setFlag(CDKConstants.ISAROMATIC, true);
bond3.setFlag(CDKConstants.ISAROMATIC, true);
bond4.setFlag(CDKConstants.ISAROMATIC, true);
bond5.setFlag(CDKConstants.ISAROMATIC, true);
bond6.setFlag(CDKConstants.ISAROMATIC, true);
Electron counts
Bond orders, as we have seen earlier, are commonly used in the CDK to indicate the electronic properties of a bond. At the same time, each bond consists of a number of atoms. For example, in a single (sigma) bond, two electrons are involved. In a double (pi) bond, four electrons are involved, and in a triple bond, six electrons are involved. We can report on the electron counts for the various orders with this code:
Script 3.13 code/ElectronCounts.groovy
IBond.Order.each { order ->
bond = new Bond(
new Atom("C"), new Atom("C"),
order
)
println "Bond order $order has " +
bond.electronCount + " electrons"
}
showing us the default implementation:
Bond order SINGLE has 2 electrons
Bond order DOUBLE has 4 electrons
Bond order TRIPLE has 6 electrons
Bond order QUADRUPLE has 8 electrons
Bond order QUINTUPLE has 10 electrons
Bond order SEXTUPLE has 12 electrons
Bond order UNSET has 0 electrons
Bond stereochemistry
The IBond.setStereo() method is discussed in Section 5.1.
Molecules
We already saw in the previous pieces of code how the CDK can be used to create molecules, and while the above is, strictly speaking, enough to find all atoms in the molecule starting with only one of the atoms in the molecule, it often is more convenient to store all atoms and bonds in a container.
The CDK has one container: the IAtomContainer.
It is a general container to holds atoms an bonds, and can contain both
unconnected as well asfully connected structures. The latter
has the added implication that it holds a single molecule, of which all
atoms are connected to each other via one or more covalent bonds.
Adding atoms and bonds is done by the methods addAtom(IAtom) and
addBond(IBond):
Script 3.14 code/AtomContainerAddAtomsAndBonds.groovy
mol = new AtomContainer();
mol.addAtom(new Atom("C"));
mol.addAtom(new Atom("H"));
mol.addAtom(new Atom("H"));
mol.addAtom(new Atom("H"));
mol.addAtom(new Atom("H"));
mol.addBond(new Bond(mol.getAtom(0), mol.getAtom(1)));
mol.addBond(new Bond(mol.getAtom(0), mol.getAtom(2)));
mol.addBond(new Bond(mol.getAtom(0), mol.getAtom(3)));
mol.addBond(new Bond(mol.getAtom(0), mol.getAtom(4)));
The addBond() method has an alternative which takes three parameters:
the first atom, the second atom, and the bond order. Note that atom indices
follows programmers habits and starts at 0, as you can observe in the
previous example too. This shortens the previous version a bit:
Script 3.15 code/AtomContainerAddAtomsAndBonds2.groovy
mol = new AtomContainer();
mol.addAtom(new Atom("C"));
mol.addAtom(new Atom("H"));
mol.addAtom(new Atom("H"));
mol.addAtom(new Atom("H"));
mol.addAtom(new Atom("H"));
mol.addBond(0,1,IBond.Order.SINGLE);
mol.addBond(0,2,IBond.Order.SINGLE);
mol.addBond(0,3,IBond.Order.SINGLE);
mol.addBond(0,4,IBond.Order.SINGLE);
A third alternative takes advantage of the newAtom() and newBond() methods:
Script code/AtomContainerAddAtomsAndBonds3.groovy
mol = new AtomContainer();
c = mol.newAtom((int)IElement.C);
h1 = mol.newAtom((int)IElement.H);
h2 = mol.newAtom((int)IElement.H);
h3 = mol.newAtom((int)IElement.H);
h4 = mol.newAtom((int)IElement.H);
mol.newBond(c,h1,IBond.Order.SINGLE);
mol.newBond(c,h2,IBond.Order.SINGLE);
mol.newBond(c,h3,IBond.Order.SINGLE);
mol.newBond(c,h4,IBond.Order.SINGLE);
Iterating over atoms and bonds
The IAtomContainer comes with convenience methods to iterate over atoms
and bonds. Both methods use the Iterable interfaces, and for atoms we
do:
Script 3.16 code/CountHydrogens.groovy
int hydrogenCount = 0
for (IAtom atom : mol.atoms()) {
if ("H".equals(atom.getSymbol())) hydrogenCount++
}
println "Number of hydrogens: $hydrogenCount"
which returns
Number of hydrogens: 4
And for bonds the equivalent:
Script 3.17 code/CountDoubleBonds.groovy
int doubleBondCount = 0
for (IBond bond : mol.bonds()) {
if (IBond.Order.DOUBLE == bond.getOrder())
doubleBondCount++
}
println "Number of double bonds: $doubleBondCount"
giving
Number of double bonds: 1
Neighboring atoms and bonds
It is quite common that you like to see what atoms are connected
to one particular atom. For example, you may wish to count how many
bonds surround a particular atom. Or, you may want to list all atoms
that are bound to this atom. The IAtomContainer class
provides methods for these use cases. But it should be stressed that
these methods do only take into account explicit hydrogens (see the
next section).
Let’s consider ethanol again, given in Script 3.10, and count the number of neighbors for each atom:
Script 3.18 code/NeighborCount.groovy
for (atom in ethanol.atoms()) {
println atom.getSymbol() +
" " + ethanol.getConnectedAtomsCount(atom)
}
which lists for the three heavy atoms:
C 1
C 2
O 1
Similarly, we can also list all connected atoms:
Script 3.19 code/ConnectedAtoms.groovy
for (atom in ethanol.atoms()) {
print atom.getSymbol() +
" is connected to "
for (neighbor in ethanol.getConnectedAtomsList(atom)) {
print neighbor.getSymbol() + " "
}
println ""
}
which outputs:
C is connected to C
C is connected to C O
O is connected to C
We can do the same thing for connected bonds:
Script 3.20 code/ConnectedBonds.groovy
for (atom in ethanol.atoms()) {
print atom.getSymbol() +
" has bond(s)"
for (bond in ethanol.getConnectedBondsList(atom)) {
print " " + bond.getOrder()
}
println ""
}
which outputs:
C has bond(s) SINGLE
C has bond(s) SINGLE SINGLE
O has bond(s) SINGLE
Molecular Formula
Getting the molecular formula of a molecule and returning that as a String
is both done with the MolecularFormulaManipulator class:
Script 3.21 code/MFGeneration.groovy
molForm = MolecularFormulaManipulator.getMolecularFormula(
azulene
)
mfString = MolecularFormulaManipulator.getString(molForm)
println "Azulene: $mfString"
giving:
Azulene: C10H8
Implicit and Explicit Hydrogens
The CDK has two concepts for hydrogens: implicit hydrogens and explicit hydrogens. Explicit hydrogens are hydrogens that are separate vertices on the chemical graph. Implicit hydrogens, however, are not, and are attributes of existing vertices.

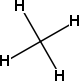
Figure 4.2: Methane with implicit (left) and explicit (right) hydrogens.
For example, if we represent methane as a chemical graph, we can define either a hydrogen-depleted chemical graph with a single carbon atom and zero bonds, or a graph with one carbon and four hydrogen atoms, and four bonds connecting the hydrogens to the central carbon. In the latter case, the hydrogens are explicit, while in the former case we can add those four hydrogens as implicit hydrogens on these carbon.
The first option in CDK code looks like:
Script 3.24 code/HydrogenDepletedGraph.groovy
molecule = new AtomContainer();
carbon = new Atom(Elements.CARBON);
carbon.setImplicitHydrogenCount(4);
molecule.addAtom(carbon);
while the alternative look like:
Script 3.25 code/HydrogenExplicitGraph.groovy
molecule = new AtomContainer();
carbon = new Atom(Elements.CARBON);
molecule.addAtom(carbon);
for (int i=1; i<=4; i++) {
hydrogen = new Atom(Elements.HYDROGEN);
molecule.addAtom(hydrogen);
molecule.addBond(0, i, IBond.Order.SINGLE);
}
Section 15.4 describes how hydrogens can be added programmatically.
Chemical Objects
Another interface that must be introduced is the IChemOject
as it plays an key role in the CDK data model. Almost all interfaces
used in the data model inherit from this interface. The IChemObject
interface provides a bit of basic functionality, including support
for object identifiers, properties, and flags.
For example. identifiers are set and retrieved with the setID() and
getID() methods:
Script 3.26 code/ChemObjectIdentifiers.groovy
butane = new AtomContainer();
butane.setID("cdkbook000000001")
print "ID: " + butane.getID()
If you have more than one identifier, or other properties you like to
associate with objects, you can use the setProperty() and
getProperty() methods:
Script 3.27 code/ChemObjectProperties.groovy
butane = new AtomContainer();
butane.setProperty(
"InChI", "InChI=1/C4H10/c1-3-4-2/h3-4H2,1-2H3"
)
print "InChI: " + butane.getProperty("InChI")
For example, we can use this approach to assign labels to atoms, such as in this example from substructure searching (see Chapter ??):
Script 3.28 code/AtomLabels.groovy
butane = MoleculeFactory.makeAlkane(4);
butane.atoms().each { atom ->
atom.setProperty("Label", "Molecule")
}
ccc = MoleculeFactory.makeAlkane(3);
ccc.atoms().each { atom ->
atom.setProperty("Label", "Substructure")
}
The CDKConstants class provides a few constants for common properties:
Script 3.29 code/CDKConstantsProperties.groovy
println "Title: " +
aspirin.getProperty(CDKConstants.TITLE)
println "InChI: " +
aspirin.getProperty(CDKConstants.INCHI)
println "SMILES: " +
aspirin.getProperty(CDKConstants.SMILES)
println "CAS registry number: " +
aspirin.getProperty(CDKConstants.CASRN)
println "COMMENT: " +
aspirin.getProperty(CDKConstants.COMMENT)
println "NAMES: " +
aspirin.getProperty(CDKConstants.NAMES)
outputting:
Title: aspirin
InChI: InChI=1/C9H8O4/c1-6(10)13-8-5-3-2-4-7(8)9(11)12/h2-5H,1H3,(H,11,12)
SMILES: CC(=O)Oc1ccccc1C(=O)O
CAS registry number: 50-78-2
COMMENT: Against headaches.
NAMES: 2-(acetyloxy)benzoic acid
A third characteristic of the IChemObject interface is the concept of
flags. Flags are used in the CDK to indicate, for example, if
an atom or bond is aromatic (see Script 3.12)
or if an atom is part of a ring:
Script 3.30 code/RingBond.groovy
benzene = MoleculeFactory.makeBenzene();
benzene.bonds().each { bond ->
bond.setFlag(CDKConstants.ISINRING, true)
println "Is ring bond: " +
bond.getFlag(CDKConstants.ISINRING)
}
The next section talks about the CDK data class for \topic{rings}.
Rings
One important aspect of molecules is rings, partly because rings can show interesting chemical phenomena. For example, if the number of FIXME electrons is right, then the ring will become aromatic, as we commonly observer in phenyl rings, such as in benzene. But, cheminformatics has many other aspects where one like to know about those rings. For example, 2D coordinate generator (see Section 15.5) requires algorithms to know what the rings are in a molecule.
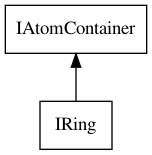
Figure 4.3: The IRing interface extends the IAtomContainer interface and is used to hold information about rings.
Section 14.2 explains what functionality the CDK has to
determine a bond takes part in a ring system. Here, we just introduce the
IRing interface, which extends the more general IAtomContainer
as shown in Figure 4.3. Practically, there is nothing much to
say about the IRing interface. One method it adds, is to get the size of the
ring:
Script 3.31 code/RingExample.groovy
IRing ring = new Ring(5, "C")
println "Ring size: " + ring.getRingSize()
println "Ring atoms: " + ring.getAtomCount()
println "Ring bonds: " + ring.getBondCount()
But this should be by definition the same as the number as atoms and bonds:
Ring size: 5
Ring atoms: 5
Ring bonds: 5
An overview of three algorithms to find rings in atom containers is provided in Section 14.3. Additionally, you may also be interested in ring sets, explained in Section ??.
References
- Steinbeck C, Han Y, Kuhn S, Horlacher O, Luttmann E, Willighagen E. The Chemistry Development Kit (CDK): an open-source Java library for Chemo- and Bioinformatics. JCICS. 2003 Feb 11;43(2):493–500. doi:10.1021/CI025584Y (Scholia)
- Balaban AT. Applications of graph theory in chemistry. JCICS. 1985 Aug 1;25(3):334–43. doi:10.1021/CI00047A033 (Scholia)