Making Computational Workflows FAIR¶

Main Objectives¶
The main purpose of this recipe is:
Provide guidance on resources available to help developers and data scientists make the various workflows used for daily tasks (for extract-load-transform, quality control, deployment or analytical workflow) available in open format and reusable.
Provide guidance for regulatory submissions for nucleic acid sequence analysis using the BioCompute Object (BCO) specification.
Remind the active nature of the field and the fast evolving environment and platforms developed for these tasks.
Provide an example using the Apache Airflow framework to illustrate the process.
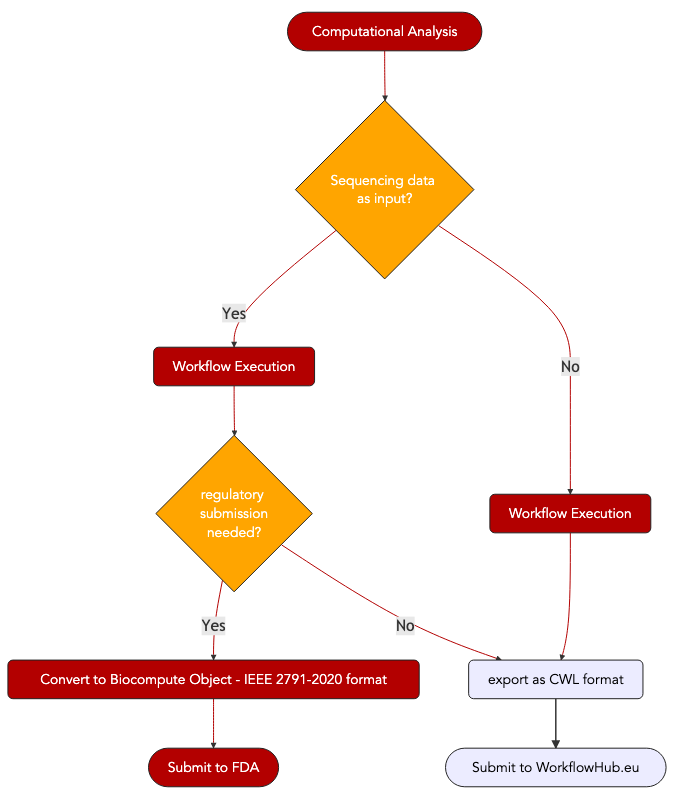
FAIRification Objectives, Inputs and Outputs¶
Actions.Objectives.Tasks |
Input |
Output |
|---|---|---|
Table of Data Standards¶
Data Formats |
Terminologies |
Models |
|---|---|---|
Tools:¶
Name |
URL |
type |
|---|---|---|
Apache Airflow |
workflow engine |
|
Galaxy |
https://galaxy.aws.biochemistry.gwu.edu/root/login?redirect=%2F |
workflow engine |
Hive |
workflow engine |
|
BioCompute Platform |
workflow engine |
|
SevenBridges BioCompute App |
workflow engine |
|
CWL-Airflow |
adapter |
Main Content¶
Workflows are ubiquitous in the data science ecosystem. The ability to automate repetitive tasks to build complex pipelines, schedule and distribute tasks to cloud infrastructures have popularized the use of workflow engine and somehow contributing to reducing the risk of errors associated with human operator fatigue. Workflow engines such as Galaxy 2, Snakemake5, Cromwell7, Knime3, Apache Airflow1, and Toil 6 to name a few offerings, have popularized the use of workflows in the field of life science computational applications. This however be can also become a source of difficulty when buying-in in a particular platform and then trying to exchange information with other platforms or migration away from the initial choice. Hence, a community of experts has dedicated efforts to define open specifications for the description of workflows as well as supporting tools, such as converters.
Using an example based on Next Generation Sequencing (NGS) application, the present content will show the reader how to
make workflow more interoperable and reusable thanks to the use of existing, off-the-shelf tools.
1. CWL: Common Workflow Language - A brief overview¶
CWL, short for Common Workflow Language, is an open standard developed by a consortium of experts, including workflow engine developers, data scientists, data analysts and bioinformaticians.
CWL specifications are available from: https://www.commonwl.org/v1.2/Workflow.html
CWL use YAML syntax to describe workflow steps, tools, input, output and parameters.
CWL is meant to provide for platform-independent workflow description, meaning that people should ideally describe workflows once to be able to execute them on CWL aware workflow engines.
CWL is currently implemented by an increasing number of platforms, which are listed here
CWL user guide is available here: http://www.commonwl.org/user_guide/
See a CWL example
cwlVersion: v1.0
class: Workflow
requirements:
- class: SubworkflowFeatureRequirement
- class: ScatterFeatureRequirement
- class: StepInputExpressionRequirement
- class: InlineJavascriptRequirement
- class: MultipleInputFeatureRequirement
inputs:
indices_folder:
type: Directory
label: "BOWTIE indices folder"
doc: "Path to BOWTIE generated indices folder"
annotation_file:
type: File
label: "Annotation file"
format: "http://edamontology.org/format_3475"
doc: "Tab-separated input annotation file"
genome_size:
type: string
label: "Effective genome size"
doc: "MACS2 effective genome size: hs, mm, ce, dm or number, for example 2.7e9"
chrom_length:
type: File
label: "Chromosome length file"
format: "http://edamontology.org/format_2330"
doc: "Chromosome length file"
control_file:
type: File?
default: null
label: "Control BAM file"
format: "http://edamontology.org/format_2572"
doc: "Control BAM file file for MACS2 peak calling"
fastq_file:
type: File
label: "FASTQ input file"
format: "http://edamontology.org/format_1930"
doc: "Reads data in a FASTQ format, received after single end sequencing"
...
remove_duplicates:
type: boolean?
default: false
label: "Remove duplicates"
doc: "Calls samtools rmdup to remove duplicates from sortesd BAM file"
threads:
type: int?
default: 2
doc: "Number of threads for those steps that support multithreading"
label: "Number of threads"
outputs:
bigwig:
type: File
format: "http://edamontology.org/format_3006"
label: "BigWig file"
doc: "Generated BigWig file"
outputSource: bam_to_bigwig/bigwig_file
fastx_statistics:
type: File
label: "FASTQ statistics"
format: "http://edamontology.org/format_2330"
doc: "fastx_quality_stats generated FASTQ file quality statistics file"
outputSource: fastx_quality_stats/statistics_file
bowtie_log:
type: File
label: "BOWTIE alignment log"
format: "http://edamontology.org/format_2330"
doc: "BOWTIE generated alignment log"
outputSource: bowtie_aligner/log_file
steps:
extract_fastq:
run: ./tools/extract-fastq.cwl
in:
compressed_file: fastq_file
out: [fastq_file]
fastx_quality_stats:
run: ./tools/fastx-quality-stats.cwl
in:
input_file: extract_fastq/fastq_file
out: [statistics_file]
2. Conditional Workflow and the CWL when keyword:¶
When describing a protocol, it is often desirable to what to do if a specific situation arises. Computational workflows are no different, and it is in fact quite frequent to have the need to define specific sets of steps if a threshold or condition is met. Therefore, the Common Workflow Language contains a dedicated keyword when to represent such situations. The following block shows how it can be used with a example:
http://www.commonwl.org/user_guide/24_conditional-workflow/index.html
class: Workflow
cwlVersion: v1.2
inputs:
val: int
steps:
step1:
in:
in1: val
a_new_var: val
run: foo.cwl
when: $(inputs.in1 < 1)
out: [out1]
step2:
in:
in1: val
a_new_var: val
run: foo.cwl
when: $(inputs.a_new_var > 2)
out: [out1]
outputs:
out1:
type: string
outputSource:
- step1/out1
- step2/out1
pickValue: first_non_null
requirements:
InlineJavascriptRequirement: {}
MultipleInputFeatureRequirement: {}
3. Semantic Markup of CWL workflows¶
CWL documents can be annotated with Schema.org or EDAM vocabulary elements to support findability.
The blocks of code below shows how this is done with 2 examples.
#!/usr/bin/env cwl-runner
cwlVersion: v1.0
class: CommandLineTool
label: An example tool demonstrating metadata.
doc: Note that this is an example and the metadata is not necessarily consistent.
hints:
ResourceRequirement:
coresMin: 4
inputs:
aligned_sequences:
type: File
label: Aligned sequences in BAM format
format: edam:format_2572
inputBinding:
position: 1
baseCommand: [ wc, -l ]
stdout: output.txt
outputs:
report:
type: stdout
format: edam:format_1964
label: A text file that contains a line count
s:author:
- class: s:Person
s:identifier: https://orcid.org/0000-0002-6130-1021
s:email: mailto:dyuen@oicr.on.ca
s:name: Denis Yuen
s:contributor:
- class: s:Person
s:identifier: http://orcid.org/0000-0002-7681-6415
s:email: mailto:briandoconnor@gmail.com
s:name: Brian O'Connor
s:citation: https://dx.doi.org/10.6084/m9.figshare.3115156.v2
s:codeRepository: https://github.com/common-workflow-language/common-workflow-language
s:dateCreated: "2016-12-13"
s:license: https://spdx.org/licenses/Apache-2.0
s:keywords: edam:topic_0091 , edam:topic_0622
s:programmingLanguage: C
$namespaces:
s: https://schema.org/
edam: http://edamontology.org/
$schemas:
- https://schema.org/version/latest/schemaorg-current-http.rdf
- http://edamontology.org/EDAM_1.18.owl
**View another example
$namespaces:
s: http://schema.org/
$schemas:
- http://schema.org/docs/schema_org_rdfa.html
s:name: "biowardrobe_chipseq_se"
s:downloadUrl: https://raw.githubusercontent.com/Barski-lab/ga4gh_challenge/master/biowardrobe_chipseq_se.cwl
s:codeRepository: https://github.com/Barski-lab/ga4gh_challenge
s:license: http://www.apache.org/licenses/LICENSE-2.0
s:isPartOf:
class: s:CreativeWork
s:name: Common Workflow Language
s:url: http://commonwl.org/
s:creator:
- class: s:Organization
s:legalName: "Cincinnati Children's Hospital Medical Center"
s:location:
- class: s:PostalAddress
s:addressCountry: "USA"
s:addressLocality: "Cincinnati"
s:addressRegion: "OH"
s:postalCode: "45229"
s:streetAddress: "3333 Burnet Ave"
s:telephone: "+1(513)636-4200"
s:logo: "https://www.cincinnatichildrens.org/-/media/cincinnati%20childrens/global%20shared/childrens-logo-new.png"
s:department:
- class: s:Organization
s:legalName: "Allergy and Immunology"
s:department:
- class: s:Organization
s:legalName: "Barski Research Lab"
s:member:
- class: s:Person
s:name: Michael Kotliar
s:email: mailto:misha.kotliar@gmail.com
s:sameAs:
- id: http://orcid.org/0000-0002-6486-3898
doc: |
The workflow is used to run CHIP-Seq basic analysis with single-end input FASTQ file.
In outputs it returns coordinate sorted BAM file alongside with index BAI file, quality
statistics of the input FASTQ file, reads coverage in a form of bigWig file, peaks calling
data in a form of narrowPeak or broadPeak files.
s:about: |
The workflow is a CWL version of a Python pipeline from BioWardrobe (Kartashov and Barski, 2015).
It starts by extracting input FASTQ file (in case it was compressed). Next step runs BowTie
(Langmead et al., 2009) to perform alignment to a reference genome, resulting in an unsorted SAM file.
The SAM file is then sorted and indexed with samtools (Li et al., 2009) to obtain a BAM file and a BAI index.
Next MACS2 (Zhang et al., 2008) is used to call peaks and to estimate fragment size. In the last few steps,
the coverage by estimated fragments is calculated from the BAM file and is reported in bigWig format. The pipeline
also reports statistics, such as read quality, peak number and base frequency, and other troubleshooting information
using tools such as fastx-toolkit and bamtools.
4. Publishing Workflows as CWL in WorkflowHub.eu:¶
Workflows are digital objects which can and should be preserved.
A number of repositories exist and may be used to deposit workflows.
One may use a generic repository such as Zenodo to do so (see recipe Depositing in Zenodo generic repository).
Preferably, one should use a specialized repository such as Workflowhub.eu, which is presented below.
Fig. 23 The european workflowhub website 1.¶
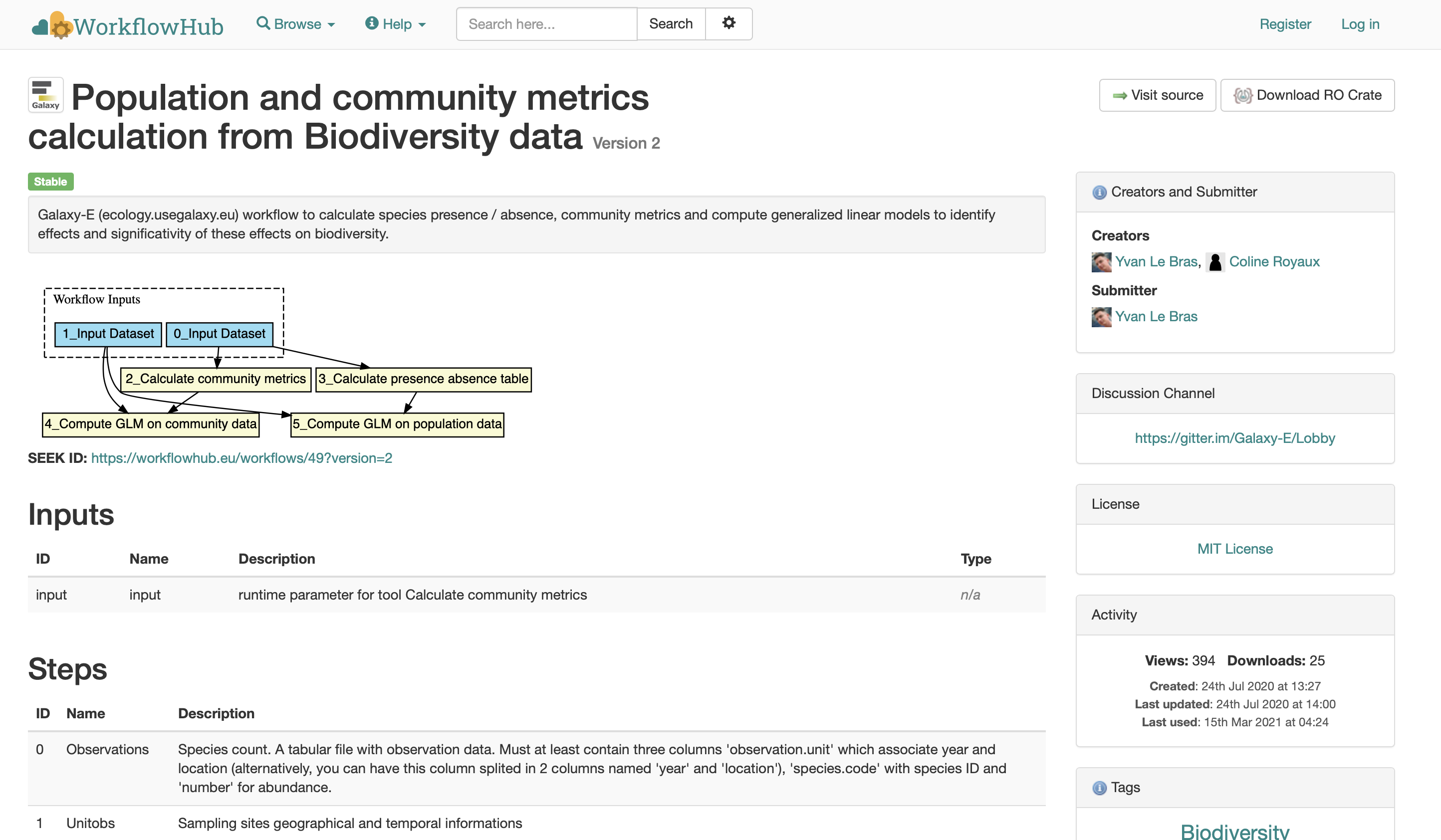
Fig. 24 The european workflowhub website 2.¶
5. Tools: Apache AIRflow playing with CWL¶
Apache Airflow is a platform created by the community to programmatically author, schedule and monitor workflows , to quote the project’s site. It has established itself in industry settings and has broad uptake.
Apache Airflow represents workflows as Directed Acyclic Graph (or DAGs) and Airflow allows the serialization of these as JSON documents.
The main thing about Apache Airflow is that code is used to generate the workflows. For more information, refer to this tutorial: https://airflow.apache.org/docs/apache-airflow/stable/tutorial.html.
A tool developed by Michael Kotliar, Andrey V Kartashov, Artem Barski brings CWL support to the Apache Airflow framework, meaning that CWL expressed workflow can now be executed on the platform 4.
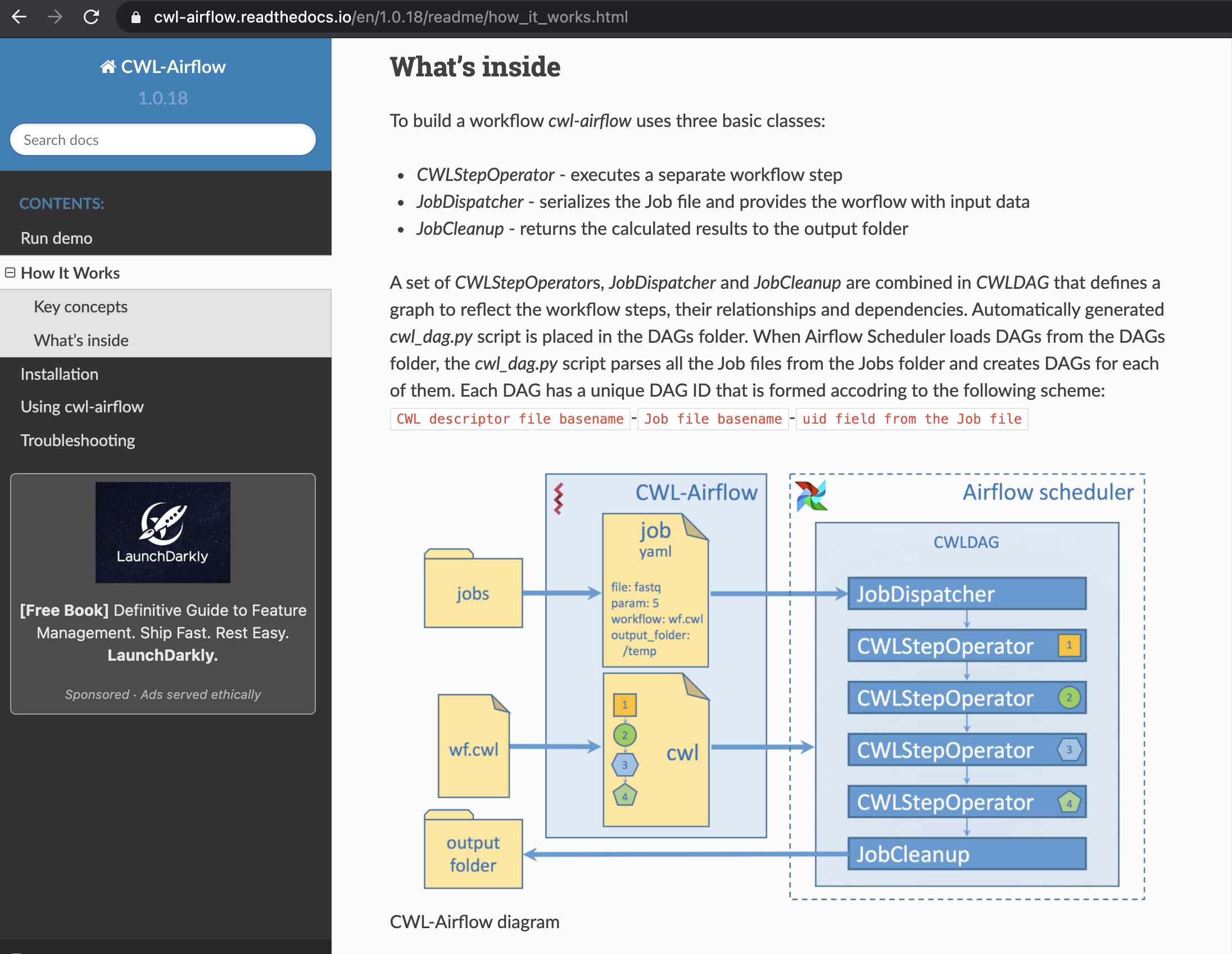
Fig. 25 the CWL-Airflow component.¶
A key step in this linkage is the conversion of a CWL expressed workflow into an Apache Airflow DAG, which can then be subsequently executed.
With this example, we aim to bring awareness about the value of having platform independent expression of workflows.
6. Biocompute Object format, an IEEE specification suited for use in regulatory applications.¶
If computational analyses on sequence data are performed in the context of clinical trials, for instance to demonstrate the transcriptomics response to a drug or to show to safety of a compound in populations of distinct genetic background using genotyping information, it is a regulatory requirements of the US FDA to submit the computational workflows if seeking approval. The availability of such information in this context is a prerequisite for FDA auditors to examine the data.
The IEEE 2791-2020 specifications, also known as BCO for BioCompute Object is a specification to do this.
This has been made possible thanks to the fast-track submission of a new data format specifically tailored to ensure reproducibility and unambiguous description of workflow key descriptors.

Fig. 26 Cloud based tools supported BCO specifications¶
What are the main features of a BioCompute Object?¶
a BioCompute Object is serialized as a JSON document. A typical BCO looks like this:
{
"object_id": "urn:uuid:dc308d7c-7949-446a-9c39-511b8ab40caf",
"spec_version": "https://w3id.org/ieee/ieee-2791-schema/",
"etag": "f8b213e62dfc7d05934ffdb7a36e4661f13b9cd04ad2de3ff3da6e933c4aebc8",
"provenance_domain": {
"name": "nf-core/chipseq: ChIP-seq peak-calling, QC and differential analysis pipeline",
"version": "1.2.1",
"created": "2020-07-29T17:33:46+01:00",
"modified": "2020-09-10T13:11:58+01:00",
"contributors": [
{"contribution": ["authoredBy"], "name": "Harshil Patel" },
{"contribution": ["authoredBy"], "name": "Chuan Wang" },
{"contribution": ["authoredBy"], "name": "Phil Ewels" },
{"contribution": ["authoredBy"], "name": "Alexander Peltzer" },
{"contribution": ["authoredBy"], "name": "Tiago Chedraoui Silva" },
{"contribution": ["authoredBy"], "name": "Drew Behrens" },
{"contribution": ["authoredBy"], "name": "Maxime Garcia" },
{"contribution": ["authoredBy"], "name": "mashehu" },
{"contribution": ["authoredBy"], "name": "Rotholandus" },
{"contribution": ["authoredBy"], "name": "Sofia Haglund" },
{"contribution": ["authoredBy"], "name": "Winni Kretzschmar" },
{"contribution": ["createdBy"],
"name": "Stian Soiland-Reyes",
"orcid": "https://orcid.org/0000-0001-9842-9718"
}
],
"license": "https://github.com/nf-core/chipseq/blob/1.2.1/LICENSE"
},
"usability_domain": [
"nfcore/chipseq is a bioinformatics analysis pipeline used for Chromatin ImmunopreciPitation sequencing (ChIP-seq) data.",
"For use with multiple replicates, the group identifier should be identical when you have multiple replicates from the same experimental group, just increment the replicate identifier appropriately. The first replicate value for any given experimental group must be 1.",
"Both the group and replicate identifiers should be the same when you have re-sequenced the same sample more than once e.g. to increase sequencing depth. The pipeline will perform the alignments in parallel, and subsequently merge them before further analysis. "
],
"description_domain": {
"keywords": [
"chipseq",
"QC"
],
"pipeline_steps": [
{"step_number": 1, "name": "CHECK_DESIGN", "description": "", "input_list": [], "output_list": []},
{"step_number": 2, "name": "output_documentation", "description": "", "input_list": [], "output_list": []},
{"step_number": 3, "name": "MAKE_GENE_BED", "description": "", "input_list": [], "output_list": []},
{"step_number": 5, "name": "get_software_versions", "description": "", "input_list": [], "output_list": []},
{"step_number": 6, "name": "BWA_INDEX", "description": "", "input_list": [], "output_list": []},
{"step_number": 7, "name": "MAKE_GENOME_FILTER", "description": "", "input_list": [], "output_list": []},
{"step_number": 8, "name": "FASTQC", "description": "", "input_list": [], "output_list": []},
{"step_number": 9, "name": "TRIMGALORE", "description": "", "input_list": [], "output_list": []},
{"step_number": 10, "name": "BWA_MEM", "description": "", "input_list": [], "output_list": []},
{"step_number": 11, "name": "SORT_BAM", "description": "", "input_list": [], "output_list": []},
{"step_number": 12, "name": "MERGED_BAM", "description": "", "input_list": [], "output_list": []},
{"step_number": 13, "name": "PRESEQ", "description": "", "input_list": [], "output_list": []},
{"step_number": 14, "name": "MERGED_BAM_FILTER", "description": "", "input_list": [], "output_list": []},
{"step_number": 15, "name": "MERGED_BAM_REMOVE_ORPHAN", "description": "", "input_list": [], "output_list": []},
{"step_number": 16, "name": "PHANTOMPEAKQUALTOOLS", "description": "", "input_list": [], "output_list": []},
{"step_number": 17, "name": "BIGWIG", "description": "", "input_list": [], "output_list": []},
{"step_number": 18, "name": "PICARD_METRICS", "description": "", "input_list": [], "output_list": []},
{"step_number": 19, "name": "PLOTFINGERPRINT", "description": "", "input_list": [], "output_list": []},
{"step_number": 20, "name": "MACS2", "description": "", "input_list": [], "output_list": []},
{"step_number": 21, "name": "PLOTPROFILE", "description": "", "input_list": [], "output_list": []},
{"step_number": 22, "name": "MACS2_ANNOTATE", "description": "", "input_list": [], "output_list": []},
{"step_number": 23, "name": "CONSENSUS_PEAKS", "description": "", "input_list": [], "output_list": []},
{"step_number": 24, "name": "CONSENSUS_PEAKS_COUNTS", "description": "", "input_list": [], "output_list": []},
{"step_number": 25, "name": "CONSENSUS_PEAKS_ANNOTATE", "description": "", "input_list": [], "output_list": []},
{"step_number": 26, "name": "MACS2_QC", "description": "", "input_list": [], "output_list": []},
{"step_number": 27, "name": "CONSENSUS_PEAKS_DESEQ2", "description": "", "input_list": [], "output_list": []},
{"step_number": 28, "name": "MULTIQC", "description": "", "input_list": [], "output_list": []},
{"step_number": 29, "name": "IGV", "description": "", "input_list": [], "output_list": []},
{"step_number": 30, "name": "but", "description": "", "input_list": [], "output_list": []},
{"step_number": 31, "name": "errored", "description": "", "input_list": [], "output_list": []},
{"step_number": 32, "name": "ran", "description": "", "input_list": [], "output_list": []}
]
},
"execution_domain": {
"script": ["https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"],
"script_driver": "nextflow",
"software_prerequisites": [
{"name": "nextflow",
"version": "20.07",
"uri": {
"uri": "http://nextflow.io/"
}
},
{ "name": "conda",
"version": "4.8.4",
"uri": {
"uri": "https://docs.conda.io/en/latest/miniconda.html"
}
},
{"name": "python", "version": "3.7.6", "uri": {"uri": "https://anaconda.org/conda-forge/python"} },
{"name": "markdown", "version": "3.2.2", "uri": {"uri": "https://anaconda.org/conda-forge/markdown"} },
{"name": "pymdown-extensions", "version": "7.1", "uri": {"uri": "https://anaconda.org/conda-forge/pymdown-extensions"} },
{"name": "pygments", "version": "2.6.1", "uri": {"uri": "https://anaconda.org/conda-forge/pygments"} },
{"name": "r-base", "version": "3.6.2", "uri": {"uri": "https://anaconda.org/conda-forge/r-base"} },
{"name": "r-optparse", "version": "1.6.6", "uri": {"uri": "https://anaconda.org/conda-forge/r-optparse"} },
{"name": "r-rcolorbrewer", "version": "1.1_2", "uri": {"uri": "https://anaconda.org/conda-forge/r-rcolorbrewer"} },
{"name": "r-reshape2", "version": "1.4.4", "uri": {"uri": "https://anaconda.org/conda-forge/r-reshape2"} },
{"name": "r-ggplot2", "version": "3.3.2", "uri": {"uri": "https://anaconda.org/conda-forge/r-ggplot2"} },
{"name": "r-tidyr", "version": "1.1.0", "uri": {"uri": "https://anaconda.org/conda-forge/r-tidyr"} },
{"name": "r-scales", "version": "1.1.1", "uri": {"uri": "https://anaconda.org/conda-forge/r-scales"} },
{"name": "r-pheatmap", "version": "1.0.12", "uri": {"uri": "https://anaconda.org/conda-forge/r-pheatmap"} },
{"name": "r-lattice", "version": "0.20_41", "uri": {"uri": "https://anaconda.org/conda-forge/r-lattice"} },
{"name": "r-upsetr", "version": "1.4.0", "uri": {"uri": "https://anaconda.org/conda-forge/r-upsetr"} },
{"name": "r-xfun", "version": "0.15", "uri": {"uri": "https://anaconda.org/conda-forge/r-xfun"} },
{"name": "gawk", "version": "5.1.0", "uri": {"uri": "https://anaconda.org/conda-forge/gawk"} },
{"name": "pigz", "version": "2.3.4", "uri": {"uri": "https://anaconda.org/conda-forge/pigz"} },
{"name": "fastqc", "version": "0.11.9", "uri": {"uri": "https://bioconda.github.io/recipes/fastqc/README.html"} },
{"name": "trim-galore", "version": "0.6.5", "uri": {"uri": "https://bioconda.github.io/recipes/trim-galore/README.html"} },
{"name": "bwa", "version": "0.7.17", "uri": {"uri": "https://bioconda.github.io/recipes/bwa/README.html"} },
{"name": "samtools", "version": "1.10", "uri": {"uri": "https://bioconda.github.io/recipes/samtools/README.html"} },
{"name": "picard", "version": "2.23.1", "uri": {"uri": "https://bioconda.github.io/recipes/picard/README.html"} },
{"name": "bamtools", "version": "2.5.1", "uri": {"uri": "https://bioconda.github.io/recipes/bamtools/README.html"} },
{"name": "pysam", "version": "0.15.3", "uri": {"uri": "https://bioconda.github.io/recipes/pysam/README.html"} },
{"name": "bedtools", "version": "2.29.2", "uri": {"uri": "https://bioconda.github.io/recipes/bedtools/README.html"} },
{"name": "ucsc-bedgraphtobigwig", "version": "357", "uri": {"uri": "https://bioconda.github.io/recipes/ucsc-bedgraphtobigwig/README.html"} },
{"name": "deeptools", "version": "3.4.3", "uri": {"uri": "https://bioconda.github.io/recipes/deeptools/README.html"} },
{"name": "macs2", "version": "2.2.7.1", "uri": {"uri": "https://bioconda.github.io/recipes/macs2/README.html"} },
{"name": "homer", "version": "4.11", "uri": {"uri": "https://bioconda.github.io/recipes/homer/README.html"} },
{"name": "subread", "version": "2.0.1", "uri": {"uri": "https://bioconda.github.io/recipes/subread/README.html"} },
{"name": "phantompeakqualtools", "version": "1.2.2", "uri": {"uri": "https://bioconda.github.io/recipes/phantompeakqualtools/README.html"} },
{"name": "preseq", "version": "2.0.3", "uri": {"uri": "https://bioconda.github.io/recipes/preseq/README.html"} },
{"name": "multiqc", "version": "1.9", "uri": {"uri": "https://bioconda.github.io/recipes/multiqc/README.html"} },
{"name": "bioconductor-biocparallel", "version": "1.20.0", "uri": {"uri": "https://bioconda.github.io/recipes/bioconductor-biocparallel/README.html"} },
{"name": "bioconductor-deseq2", "version": "1.26.0", "uri": {"uri": "https://bioconda.github.io/recipes/bioconductor-deseq2/README.html"} },
{"name": "bioconductor-vsn", "version": "3.54.0", "uri": {"uri": "https://bioconda.github.io/recipes/bioconductor-vsn/README.html"} }
],
"external_data_endpoints": [
{"name": "Experiment design file for minimal test dataset",
"url": "https://raw.githubusercontent.com/nf-core/test-datasets/chipseq/design.csv"
},
{"name": "iGenomes R64-1-1 Ensembl (Fasta sequence)",
"url": "https://raw.githubusercontent.com/nf-core/test-datasets/atacseq/reference/genome.fa"
},
{"name": "iGenomes R64-1-1 Ensembl (GTF Genes)",
"url": "https://raw.githubusercontent.com/nf-core/test-datasets/atacseq/reference/genes.gtf"
}
],
"environment_variables": {}
},
"io_domain": {
"input_subdomain": [],
"output_subdomain": []
}
}
source: https://github.com/biocompute-objects/bco-ro-example-chipseq/blob/main/data/chipseq_20200910.json
a BioCompute Object can be packaged as an RO-Crate.
View an RO-Crate json denoting a BCO
{
"@context": [
"https://w3id.org/ro/crate/1.0/context",
{
"@vocab": "https://schema.org/"
}
],
"@graph": [
{
"@id": "ro-crate-metadata.json",
"@type": "CreativeWork",
"about": {
"@id": "./"
},
"identifier": "ro-crate-metadata.json",
"conformsTo": {
"@id": "https://w3id.org/ro/crate/1.0"
},
"license": {
"@id": "https://creativecommons.org/licenses/by-sa/3.0"
},
"description": "Made with Describo: https://uts-eresearch.github.io/describo/"
},
{
"@type": "Dataset",
"author": {
"@id": "https://orcid.org/0000-0001-9842-9718"
},
"citation": {
"@id": "https://doi.org/10.5281/zenodo.3966161"
},
"contactPoint": {
"@id": "https://github.com/biocompute-objects/bco-ro-example-chipseq/issues"
},
"datePublished": "2020-09-09T23:00:00.000Z",
"description": "Workflow run of a ChIP-seq peak-calling, QC and differential analysis pipeline",
"distribution": {
"@id": "https://github.com/biocompute-objects/bco-ro-example-chipseq/archive/main.zip"
},
"hasPart": [
{
"@id": "https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"
},
{
"@id": "chipseq_20200910.json"
},
{
"@id": "results/"
},
{
"@id": "nextflow.log"
},
{
"@id": ".nextflow.log"
}
],
"license": {
"@id": "https://spdx.org/licenses/CC0-1.0"
},
"name": "Workflow run of nf-core/chipseq",
"publisher": {
"@id": "https://biocomputeobject.org/"
},
"@id": "./"
},
{
"@type": "File",
"dateModified": "2020-09-10T13:10:50.246Z",
"name": ".nextflow.log",
"@reverse": {
"hasPart": [
{
"@id": "./"
}
]
},
"@id": ".nextflow.log"
},
{
"@type": "File",
"conformsTo": {
"@id": "https://w3id.org/ieee/ieee-2791-schema/"
},
"dateModified": "2020-09-10T13:50:02.378Z",
"identifier": {
"@id": "urn:uuid:dc308d7c-7949-446a-9c39-511b8ab40caf"
},
"license": {
"@id": "https://spdx.org/licenses/CC0-1.0"
},
"name": "chipseq_20200910.json",
"description": "IEEE 2791 description",
"@reverse": {
"hasPart": [
{
"@id": "./"
}
]
},
"@id": "chipseq_20200910.json"
},
{
"@type": "Organization",
"description": " Two non-overlapping entities work in parallel to help drive BioCompute, the IEEE 2791-2020 Standard, and a Public Private Partnership. Leadership for the Public Private Partnership consists of an Executive Steering Committee and a Technical Steering Committee. The schema that is referenced by the current draft of the IEEE standard is maintained by an IEEE GitLab repository. ",
"name": "BioCompute Objects",
"@reverse": {
"publisher": [
{
"@id": "./"
}
]
},
"@id": "https://biocomputeobject.org/"
},
{
"@type": "ScholarlyArticle",
"name": "nf-core/chipseq: nf-core/chipseq v1.2.1 - Platinum Mole",
"@reverse": {
"citation": [
{
"@id": "./"
},
{
"@id": "https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"
}
]
},
"@id": "https://doi.org/10.5281/zenodo.3966161"
},
{
"@type": "CreativeWork",
"identifier": "https://spdx.org/licenses/MIT",
"name": "MIT License",
"@reverse": {
"license": [
{
"@id": "https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"
}
]
},
"@id": "https://github.com/nf-core/chipseq/blob/1.2.1/LICENSE"
},
{
"@type": "CreativeWork",
"description": "\nMIT License\n\nCopyright (c) 2018 nf-core\n\nPermission is hereby granted, free of charge, to any person obtaining a copy\nof this software and associated documentation files (the \"Software\"), to deal\nin the Software without restriction, including without limitation the rights\nto use, copy, modify, merge, publish, distribute, sublicense, and/or sell\ncopies of the Software, and to permit persons to whom the Software is\nfurnished to do so, subject to the following conditions:\n\nThe above copyright notice and this permission notice shall be included in all\ncopies or substantial portions of the Software.\n\nTHE SOFTWARE IS PROVIDED \"AS IS\", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR\nIMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY,\nFITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE\nAUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER\nLIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM,\nOUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE\nSOFTWARE.",
"name": "MIT License",
"@reverse": {
"license": [
{
"@id": "results/"
}
]
},
"@id": "https://github.com/nf-core/test-datasets/blob/atacseq/LICENSE"
},
{
"@type": "DataDownload",
"path": "https://github.com/biocompute-objects/bco-ro-example-chipseq/archive/main.zip",
"license": {
"@id": "https://spdx.org/licenses/CC0-1.0"
},
"name": "GitHub download of biocompute-objects/bco-ro-example-chipseq",
"@reverse": {
"distribution": [
{
"@id": "./"
}
]
},
"@id": "https://github.com/biocompute-objects/bco-ro-example-chipseq/archive/main.zip"
},
{
"@type": "ContactPoint",
"name": " bco-ro-example-chipseq GitHub issue tracker",
"url": "https://github.com/biocompute-objects/bco-ro-example-chipseq/issues",
"@reverse": {
"contactPoint": [
{
"@id": "./"
}
]
},
"@id": "https://github.com/biocompute-objects/bco-ro-example-chipseq/issues"
},
{
"@type": "Person",
"name": "Stian Soiland-Reyes",
"@reverse": {
"author": [
{
"@id": "./"
},
{
"@id": "results/"
}
]
},
"@id": "https://orcid.org/0000-0001-9842-9718"
},
{
"@type": [
"ComputationalWorkflow",
"File"
],
"author": [
{
"@id": "#714de175-aa77-47f1-9f99-6a4fba65530a"
},
{
"@id": "#bfb876e7-e767-4209-ad66-e1e1379c249f"
},
{
"@id": "#0164006f-bd58-4ebc-9a50-b8bd4ac3025c"
},
{
"@id": "#556c747c-376a-4a85-82a1-9b99520d24fd"
},
{
"@id": "#93c23523-03b5-41dc-be4c-6a9a2e0e221d"
},
{
"@id": "#781b9b5a-dc06-4709-8f14-65ee08b8c543"
},
{
"@id": "#f652b13e-0ba2-4394-a990-7304f54c7b9a"
},
{
"@id": "#a58abf42-751d-49bd-a477-1d5065ac70c6"
},
{
"@id": "#e11af59b-8e24-4cc8-8f5e-cef411ab0823"
},
{
"@id": "#f262954b-a218-480d-8a01-0e0b1ca20ffc"
},
{
"@id": "#64bd387d-60ad-4df8-804e-1f6b9ea72de5"
}
],
"citation": {
"@id": "https://doi.org/10.5281/zenodo.3966161"
},
"description": "nfcore/chipseq is a bioinformatics analysis pipeline used for Chromatin ImmunopreciPitation sequencing (ChIP-seq) data",
"license": {
"@id": "https://github.com/nf-core/chipseq/blob/1.2.1/LICENSE"
},
"name": "nf-core/chipseq",
"@reverse": {
"hasPart": [
{
"@id": "./"
}
],
"about": [
{
"@id": "results/pipeline_info/pipeline_dag.svg"
},
{
"@id": "#fcb32545-04bd-474d-9b6e-0fb7321c38b4"
}
]
},
"@id": "https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"
},
{
"@type": "File",
"creator": {
"@id": "#db65dfb7-4867-400e-a12f-a1652d46a333"
},
"dateModified": "2020-09-10T13:10:50.250Z",
"name": "nextflow.log",
"@reverse": {
"hasPart": [
{
"@id": "./"
}
]
},
"@id": "nextflow.log"
},
{
"@type": "Dataset",
"author": {
"@id": "https://orcid.org/0000-0001-9842-9718"
},
"creator": {
"@id": "#db65dfb7-4867-400e-a12f-a1652d46a333"
},
"dateModified": "2020-09-10T13:20:49.143Z",
"description": "Nextflow outputs from examplar run of nf-core/ pipeline workflow.",
"hasPart": [
{
"@id": "results/bwa/"
},
{
"@id": "results/fastqc/"
},
{
"@id": "results/genome/"
},
{
"@id": "results/igv/"
},
{
"@id": "results/multiqc/"
},
{
"@id": "results/pipeline_info/"
},
{
"@id": "results/trim_galore/"
}
],
"license": {
"@id": "https://github.com/nf-core/test-datasets/blob/atacseq/LICENSE"
},
"name": "results",
"@reverse": {
"hasPart": [
{
"@id": "./"
}
]
},
"@id": "results/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T12:00:09.238Z",
"hasPart": {
"@id": "results/bwa/mergedLibrary/"
},
"name": "bwa",
"@reverse": {
"hasPart": [
{
"@id": "results/"
}
]
},
"@id": "results/bwa/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T12:02:59.495Z",
"hasPart": [
{
"@id": "results/bwa/mergedLibrary/bigwig/"
},
{
"@id": "results/bwa/mergedLibrary/deepTools/"
},
{
"@id": "results/bwa/mergedLibrary/macs/"
},
{
"@id": "results/bwa/mergedLibrary/phantompeakqualtools/"
},
{
"@id": "results/bwa/mergedLibrary/picard_metrics/"
}
],
"name": "mergedLibrary",
"@reverse": {
"hasPart": [
{
"@id": "results/bwa/"
}
]
},
"@id": "results/bwa/mergedLibrary/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T12:04:31.692Z",
"hasPart": {
"@id": "results/bwa/mergedLibrary/bigwig/scale/"
},
"name": "bigwig",
"@reverse": {
"hasPart": [
{
"@id": "results/bwa/mergedLibrary/"
}
]
},
"@id": "results/bwa/mergedLibrary/bigwig/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T12:04:31.696Z",
"name": "scale",
"@reverse": {
"hasPart": [
{
"@id": "results/bwa/mergedLibrary/bigwig/"
}
]
},
"@id": "results/bwa/mergedLibrary/bigwig/scale/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T12:11:43.943Z",
"hasPart": [
{
"@id": "results/bwa/mergedLibrary/deepTools/plotFingerprint/"
},
{
"@id": "results/bwa/mergedLibrary/deepTools/plotProfile/"
}
],
"name": "deepTools",
"@reverse": {
"hasPart": [
{
"@id": "results/bwa/mergedLibrary/"
}
]
},
"@id": "results/bwa/mergedLibrary/deepTools/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T12:05:17.700Z",
"name": "plotFingerprint",
"@reverse": {
"hasPart": [
{
"@id": "results/bwa/mergedLibrary/deepTools/"
}
]
},
"@id": "results/bwa/mergedLibrary/deepTools/plotFingerprint/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T12:26:12.375Z",
"name": "plotProfile",
"@reverse": {
"hasPart": [
{
"@id": "results/bwa/mergedLibrary/deepTools/"
}
]
},
"@id": "results/bwa/mergedLibrary/deepTools/plotProfile/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T12:02:33.471Z",
"name": "macs",
"@reverse": {
"hasPart": [
{
"@id": "results/bwa/mergedLibrary/"
}
]
},
"@id": "results/bwa/mergedLibrary/macs/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T12:04:26.336Z",
"name": "phantompeakqualtools",
"@reverse": {
"hasPart": [
{
"@id": "results/bwa/mergedLibrary/"
}
]
},
"@id": "results/bwa/mergedLibrary/phantompeakqualtools/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T12:04:45.952Z",
"name": "picard_metrics",
"@reverse": {
"hasPart": [
{
"@id": "results/bwa/mergedLibrary/"
}
]
},
"@id": "results/bwa/mergedLibrary/picard_metrics/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T11:58:56.905Z",
"hasPart": {
"@id": "results/fastqc/zips/"
},
"name": "fastqc",
"@reverse": {
"hasPart": [
{
"@id": "results/"
}
]
},
"@id": "results/fastqc/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T11:58:56.909Z",
"name": "zips",
"@reverse": {
"hasPart": [
{
"@id": "results/fastqc/"
}
]
},
"@id": "results/fastqc/zips/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T11:56:45.292Z",
"hasPart": {
"@id": "results/genome/genome.fa"
},
"name": "genome",
"@reverse": {
"hasPart": [
{
"@id": "results/"
}
]
},
"@id": "results/genome/"
},
{
"@type": "File",
"dateModified": "2020-09-10T11:56:45.324Z",
"name": "genome.fa",
"@reverse": {
"hasPart": [
{
"@id": "results/genome/"
}
]
},
"@id": "results/genome/genome.fa"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T12:26:50.263Z",
"hasPart": {
"@id": "results/igv/broadPeak/"
},
"name": "igv",
"@reverse": {
"hasPart": [
{
"@id": "results/"
}
]
},
"@id": "results/igv/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T12:26:50.267Z",
"hasPart": {
"@id": "results/igv/broadPeak/igv_session.xml"
},
"name": "broadPeak",
"@reverse": {
"hasPart": [
{
"@id": "results/igv/"
}
]
},
"@id": "results/igv/broadPeak/"
},
{
"@type": "File",
"dateModified": "2020-09-10T12:26:50.267Z",
"name": "igv_session.xml",
"@reverse": {
"hasPart": [
{
"@id": "results/igv/broadPeak/"
}
]
},
"@id": "results/igv/broadPeak/igv_session.xml"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T12:26:59.183Z",
"hasPart": {
"@id": "results/multiqc/broadPeak/"
},
"name": "multiqc",
"@reverse": {
"hasPart": [
{
"@id": "results/"
}
]
},
"@id": "results/multiqc/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T12:26:59.183Z",
"hasPart": [
{
"@id": "results/multiqc/broadPeak/multiqc_data/"
},
{
"@id": "results/multiqc/broadPeak/multiqc_report.html"
}
],
"name": "broadPeak",
"@reverse": {
"hasPart": [
{
"@id": "results/multiqc/"
}
]
},
"@id": "results/multiqc/broadPeak/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T12:26:59.207Z",
"name": "multiqc_data",
"@reverse": {
"hasPart": [
{
"@id": "results/multiqc/broadPeak/"
}
]
},
"@id": "results/multiqc/broadPeak/multiqc_data/"
},
{
"@type": "File",
"dateModified": "2020-09-10T12:26:59.191Z",
"name": "multiqc_report.html",
"@reverse": {
"hasPart": [
{
"@id": "results/multiqc/broadPeak/"
}
]
},
"@id": "results/multiqc/broadPeak/multiqc_report.html"
},
{
"@type": "Dataset",
"creator": {
"@id": "#db65dfb7-4867-400e-a12f-a1652d46a333"
},
"dateModified": "2020-09-10T12:27:01.599Z",
"hasPart": {
"@id": "results/pipeline_info/pipeline_dag.svg"
},
"name": "pipeline_info",
"@reverse": {
"hasPart": [
{
"@id": "results/"
}
]
},
"@id": "results/pipeline_info/"
},
{
"@type": [
"WorkflowSketch",
"File"
],
"about": {
"@id": "https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"
},
"dateModified": "2020-09-10T12:27:01.755Z",
"encodingFormat": "image/svg+xml",
"name": "pipeline_dag.svg",
"@reverse": {
"hasPart": [
{
"@id": "results/pipeline_info/"
}
]
},
"@id": "results/pipeline_info/pipeline_dag.svg"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T11:57:13.996Z",
"hasPart": [
{
"@id": "results/trim_galore/fastqc/"
},
{
"@id": "results/trim_galore/logs/"
}
],
"name": "trim_galore",
"@reverse": {
"hasPart": [
{
"@id": "results/"
}
]
},
"@id": "results/trim_galore/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T11:58:55.705Z",
"hasPart": {
"@id": "results/trim_galore/fastqc/zips/"
},
"name": "fastqc",
"@reverse": {
"hasPart": [
{
"@id": "results/trim_galore/"
}
]
},
"@id": "results/trim_galore/fastqc/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T11:58:55.705Z",
"name": "zips",
"@reverse": {
"hasPart": [
{
"@id": "results/trim_galore/fastqc/"
}
]
},
"@id": "results/trim_galore/fastqc/zips/"
},
{
"@type": "Dataset",
"dateModified": "2020-09-10T11:58:55.705Z",
"name": "logs",
"@reverse": {
"hasPart": [
{
"@id": "results/trim_galore/"
}
]
},
"@id": "results/trim_galore/logs/"
},
{
"@type": "Person",
"name": "Phil Ewels",
"@reverse": {
"author": [
{
"@id": "https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"
}
]
},
"@id": "#0164006f-bd58-4ebc-9a50-b8bd4ac3025c"
},
{
"@type": "CreativeWork",
"identifier": "https://spdx.org/licenses/CC0-1.0",
"name": "Creative Commons Zero v1.0 Universal",
"@reverse": {
"license": [
{
"@id": "./"
},
{
"@id": "chipseq_20200910.json"
},
{
"@id": "https://github.com/biocompute-objects/bco-ro-example-chipseq/archive/main.zip"
}
]
},
"@id": "https://spdx.org/licenses/CC0-1.0"
},
{
"@type": "Person",
"name": "Alexander Peltzer",
"@reverse": {
"author": [
{
"@id": "https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"
}
]
},
"@id": "#556c747c-376a-4a85-82a1-9b99520d24fd"
},
{
"@type": "Person",
"name": "Winni Kretzschmar",
"@reverse": {
"author": [
{
"@id": "https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"
}
]
},
"@id": "#64bd387d-60ad-4df8-804e-1f6b9ea72de5"
},
{
"@type": "Person",
"name": "Harshil Patel",
"@reverse": {
"author": [
{
"@id": "https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"
}
]
},
"@id": "#714de175-aa77-47f1-9f99-6a4fba65530a"
},
{
"@type": "Person",
"name": "Drew Behrens",
"@reverse": {
"author": [
{
"@id": "https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"
}
]
},
"@id": "#781b9b5a-dc06-4709-8f14-65ee08b8c543"
},
{
"@type": "Person",
"name": "Tiago Chedraoui Silva",
"@reverse": {
"author": [
{
"@id": "https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"
}
]
},
"@id": "#93c23523-03b5-41dc-be4c-6a9a2e0e221d"
},
{
"@type": "Person",
"name": "mashehu",
"@reverse": {
"author": [
{
"@id": "https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"
}
]
},
"@id": "#a58abf42-751d-49bd-a477-1d5065ac70c6"
},
{
"@type": "Person",
"name": "Chuan Wang",
"@reverse": {
"author": [
{
"@id": "https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"
}
]
},
"@id": "#bfb876e7-e767-4209-ad66-e1e1379c249f"
},
{
"@type": "Person",
"name": "Nextflow 19.10.0",
"@reverse": {
"creator": [
{
"@id": "nextflow.log"
},
{
"@id": "results/"
},
{
"@id": "results/pipeline_info/"
}
]
},
"@id": "#db65dfb7-4867-400e-a12f-a1652d46a333"
},
{
"@type": "Person",
"name": "Rotholandus",
"@reverse": {
"author": [
{
"@id": "https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"
}
]
},
"@id": "#e11af59b-8e24-4cc8-8f5e-cef411ab0823"
},
{
"@type": "Person",
"name": "Sofia Haglund",
"@reverse": {
"author": [
{
"@id": "https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"
}
]
},
"@id": "#f262954b-a218-480d-8a01-0e0b1ca20ffc"
},
{
"@type": "Person",
"name": "Maxime Garcia",
"@reverse": {
"author": [
{
"@id": "https://raw.githubusercontent.com/nf-core/chipseq/1.2.1/main.nf"
}
]
},
"@id": "#f652b13e-0ba2-4394-a990-7304f54c7b9a"
},
{
"@type": "PropertyValue",
"name": "object_id",
"value": "dc308d7c-7949-446a-9c39-511b8ab40caf",
"@reverse": {
"identifier": [
{
"@id": "chipseq_20200910.json"
}
]
},
"@id": "urn:uuid:dc308d7c-7949-446a-9c39-511b8ab40caf"
}
]
}
source: https://github.com/biocompute-objects/bco-ro-example-chipseq/blob/main/data/ro-crate-metadata.json
a BioCompute Object can be integrated with HL7 FHIR as a Provenance Resource.
{
"resourceType": "Provenance",
"id": "example-biocompute-object",
"text": {
"status": "generated",
"div": "<div xmlns=\"http://www.w3.org/1999/xhtml\">\n\t\t\t<p>\n\t\t\t\t<b>Generated Narrative with Details</b>\n\t\t\t</p><p>\n\t\t\t\t<b>id</b>: example-biocompute-object</p><p>\n\t\t\t\t<b>target</b>: <a href=\"http://build.fhir.org/sequence-example.html\">MolecularSequence/example</a>\n\t\t\t</p><p>\n\t\t\t\t<b>period</b>: 2017-6-6 --> (ongoing)</p><p>\n\t\t\t\t<b>recorded</b>: 2016-6-9 8:12:14</p><p>\n\t\t\t\t<b>reason</b>: antiviral resistance detection (Details: [not stated] code null = 'null', stated as\n 'antiviral resistance detection')</p>\n\t\t\t<h3>Agents</h3>\n\t\t\t<table>\n\t\t\t\t<tr>\n\t\t\t\t\t<td>-</td>\n\t\t\t\t\t<td>\n\t\t\t\t\t\t<b>Role</b>\n\t\t\t\t\t</td>\n\t\t\t\t\t<td>\n\t\t\t\t\t\t<b>Who</b>\n\t\t\t\t\t</td>\n\t\t\t\t</tr>\n\t\t\t\t<tr>\n\t\t\t\t\t<td>*</td>\n\t\t\t\t\t<td>Author (Details: http://hl7.org/fhir/provenance-participant-role code author = 'Author',\n stated as 'null')</td>\n\t\t\t\t\t<td>\n\t\t\t\t\t\t<a href=\"http://build.fhir.org/practitioner-example.html\">Practitioner/example</a>\n\t\t\t\t\t</td>\n\t\t\t\t</tr>\n\t\t\t</table>\n\t\t\t<h3>Entities</h3>\n\t\t\t<table>\n\t\t\t\t<tr>\n\t\t\t\t\t<td>-</td>\n\t\t\t\t\t<td>\n\t\t\t\t\t\t<b>Role</b>\n\t\t\t\t\t</td>\n\t\t\t\t\t<td>\n\t\t\t\t\t\t<b>Reference</b>\n\t\t\t\t\t</td>\n\t\t\t\t</tr>\n\t\t\t\t<tr>\n\t\t\t\t\t<td>*</td>\n\t\t\t\t\t<td>source</td>\n\t\t\t\t\t<td>\n\t\t\t\t\t\t<a href=\"https://hive.biochemistry.gwu.edu/cgi-bin/prd/htscsrs/servlet.cgi?pageid=bcoexample_1\">Biocompute example</a>\n\t\t\t\t\t</td>\n\t\t\t\t</tr>\n\t\t\t</table>\n\t\t</div>"
},
"target": [
{
"reference": "MolecularSequence/example"
}
],
"occurredPeriod": {
"start": "2017-06-06"
},
"recorded": "2016-06-09T08:12:14+10:00",
"activity": {
"text": "antiviral resistance detection"
},
"agent": [
{
"type": {
"coding": [
{
"system": "http://terminology.hl7.org/CodeSystem/v3-ParticipationType",
"code": "AUT"
}
]
},
"who": {
"reference": "Practitioner/example"
}
}
],
"entity": [
{
"role": "source",
"what": {
"identifier": {
"type": {
"coding": [
{
"system": "https://hive.biochemistry.gwu.edu",
"code": "biocompute",
"display": "obj.1001"
}
]
},
"value": "https://hive.biochemistry.gwu.edu/cgi-bin/prd/htscsrs/servlet.cgi?pageid=bcoexample_1"
}
}
}
]
}
a BioCompute Object may allow referencing a CWL expressed workflow thus increasing interoperability.
Conclusion¶
This recipe focused on highlighting important considerations to bear in mind when dealing with workflows as these digital objects have become essential information carriers to assist data science tasks.
While there is no shortage of tools and frameworks for building, saving, executing workflows, making sure these can be found, interpreted by machine without human intervention and executed are essential aspects of reusability and interoperability.
Data Scientists and Information managers should therefore tap into a number of standardization efforts capable of ensure appropriate provenance tracking and information preservation.
This knowledge could be harnessed to decide whether to trust the results of an analysis or a transformation process, or to decide whether to perform new ones.
What should I read next?¶
Reference¶
Reference
- 1
Apache airflow. URL: https://airflow.apache.org/.
- 2
E. Afgan, D. Baker, B. Batut, M. van den Beek, D. Bouvier, M. Cech, J. Chilton, D. Clements, N. Coraor, B. A. Grüning, A. Guerler, J. Hillman-Jackson, S. Hiltemann, V. Jalili, H. Rasche, N. Soranzo, J. Goecks, J. Taylor, A. Nekrutenko, and D. Blankenberg. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res, 46(W1):W537–W544, 07 2018.
- 3
Michael R. Berthold, Nicolas Cebron, Fabian Dill, Thomas R. Gabriel, Tobias Kötter, Thorsten Meinl, Peter Ohl, Kilian Thiel, and Bernd Wiswedel. Knime - the konstanz information miner: version 2.0 and beyond. SIGKDD Explor. Newsl., 11(1):26–31, November 2009. URL: http://doi.acm.org/10.1145/1656274.1656280, doi:10.1145/1656274.1656280.
- 4
Michael Kotliar, Andrey V Kartashov, and Artem Barski. CWL-Airflow: a lightweight pipeline manager supporting Common Workflow Language. GigaScience, 07 2019. giz084. URL: https://doi.org/10.1093/gigascience/giz084, arXiv:https://academic.oup.com/gigascience/article-pdf/8/7/giz084/28954484/giz084.pdf, doi:10.1093/gigascience/giz084.
- 5
F. Mölder, K. P. Jablonski, B. Letcher, M. B. Hall, C. H. Tomkins-Tinch, V. Sochat, J. Forster, S. Lee, S. O. Twardziok, A. Kanitz, A. Wilm, M. Holtgrewe, S. Rahmann, S. Nahnsen, and J. Köster. Sustainable data analysis with Snakemake. F1000Res, 10:33, 2021.
- 6
J. Vivian, A. A. Rao, F. A. Nothaft, C. Ketchum, J. Armstrong, A. Novak, J. Pfeil, J. Narkizian, A. D. Deran, A. Musselman-Brown, H. Schmidt, P. Amstutz, B. Craft, M. Goldman, K. Rosenbloom, M. Cline, B. O’Connor, M. Hanna, C. Birger, W. J. Kent, D. A. Patterson, A. D. Joseph, J. Zhu, S. Zaranek, G. Getz, D. Haussler, and B. Paten. Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol, 35(4):314–316, 04 2017.
- 7
Kate Voss, Geraldine Van Der Auwera, and Jeff Gentry. Full-stack genomics pipelining with gatk4 + wdl + cromwell [version 1; not peer reviewed]. 2017. URL: https://f1000research.com/slides/6-1381, doi:10.7490/f1000research.1114634.1.
Authors¶
Authors
Name |
ORCID |
Affiliation |
Type |
ELIXIR Node |
Contribution |
|---|---|---|---|---|---|
University of Oxford |
|
Writing - Original Draft |
|||
University of Oxford |
|
Writing - Review & Editing, Funding acquisition |