10. Converting from proprietary to open format¶

10.1. Main Objectives¶
Document how to convert raw data from a propriatory, vendor specific format to an open standard format.
Apply the approach to a targeted metabolic profiling using Biocrates kit produced by IMI Resolute project.
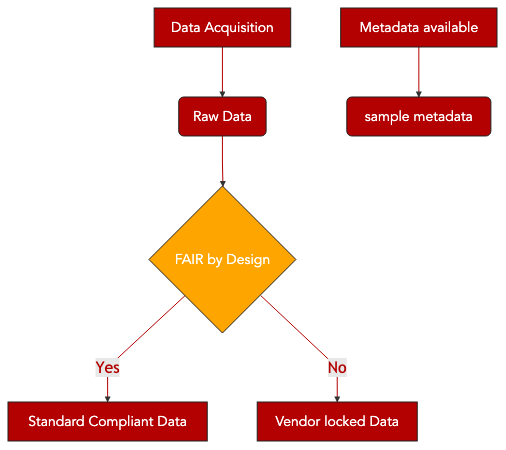
10.3. Capability & Maturity Table¶
Capability |
Initial Maturity Level |
Final Maturity Level |
|---|---|---|
Interoperability |
minimal |
repeatable |
10.4. FAIRification Objectives, Inputs and Outputs¶
Actions.Objectives.Tasks |
Input |
Output |
|---|---|---|
Waters MS format |
||
10.7. Converting Mass Spectrometry data to mzML format: a Step by Step Process.¶
10.7.1. Step 1: obtain the dataset¶
In the case of the IMI RESOLUTE project, the data is released via the University of Luxembourg server (assuming you have access resolved):
$> sftp fairplus@NNN.000.000.NNN
>get RESOLUTE_Targted_Metabolomics_of_parental_cell_lines.tar.gz
>exit
$>
:warning: NNN.000.000.NNN should be replaced with the proper IP address to the University of Luxembourg server once users have obtained data access clearance.
10.7.2. Step 2: inspect the content of the archive¶
10.7.2.1. i. copying the archive to a working directory¶
$>mv RESOLUTE_Targted_Metabolomics_of_parental_cell_lines.tar.gz /IMI-FAIR+/RESOLUTE
$>cd /IMI-FAIR+/RESOLUTE/
10.7.2.2. ii. expand the archive¶
$>gunzip RESOLUTE_Targted_Metabolomics_of_parental_cell_lines.tar.gz
$>tar -xvf RESOLUTE_Targted_Metabolomics_of_parental_cell_lines.tar
$>cd RESOLUTE_Targted_Metabolomics_of_parental_cell_lines
10.7.2.3. iii. inspect the folder¶
>$anaconda3-2019.10) bob-MacBook-3:RESOLUTE_Targted_Metabolomics_of_parental_cell_lines philippe$ ls -la
total 1400
drwxr-xr-x 10 bob staff 320 14 Jan 16:05 .
drwxr-xr-x 8 bob staff 256 14 Jan 10:29 ..
-rwxr-xr-x@ 1 bob staff 520170 15 Nov 14:02 Data_Release_Proposal_20191115_Metabolome_of_parental_cell_lines.docx
-rw-r--r--@ 1 bob staff 72868 5 Nov 20:49 EX0003_processed_data.txt
-rw-r--r--@ 1 bob staff 6907 23 Oct 16:06 EX0003_sample_metadata.tsv
drwxr-xr-x@ 7 bob staff 224 14 Jan 10:43 Raw
drwxr-xr-x 118 bob staff 3776 14 Jan 15:53 data
-rw-r--r--@ 1 bob staff 95677 7 Nov 11:02 p180_metabolites_metadata.tsv
-rw-r--r--@ 1 bob staff 162 14 Jan 10:59 ~$ta_Release_Proposal_20191115_Metabolome_of_parental_cell_lines.doc
:octopus: The archive would have benefited from having a manifest file listing all the files and their associated checksums. In so doing, it would have allowed validation and verification that no corruption happened during file transfer.
:octopus: Refer to the recipe: “How to calculate file checksums”
:octopus: Refer to the recipe: “How to package data for shipping with BDbags”
10.7.3. Step 3: Convert vendor specific format to an open format¶
One can consult the Elixir-UK FAIRsharing catalog of standards and resources to discover if an open specification exists in the domain of mass spectrometry. In this case, there is as shown below. Note that every records in the catalog has a digital object identifier (DOI), https://fairsharing.org/FAIRsharing.26dmba for HUPO-PSI mzML specifications.
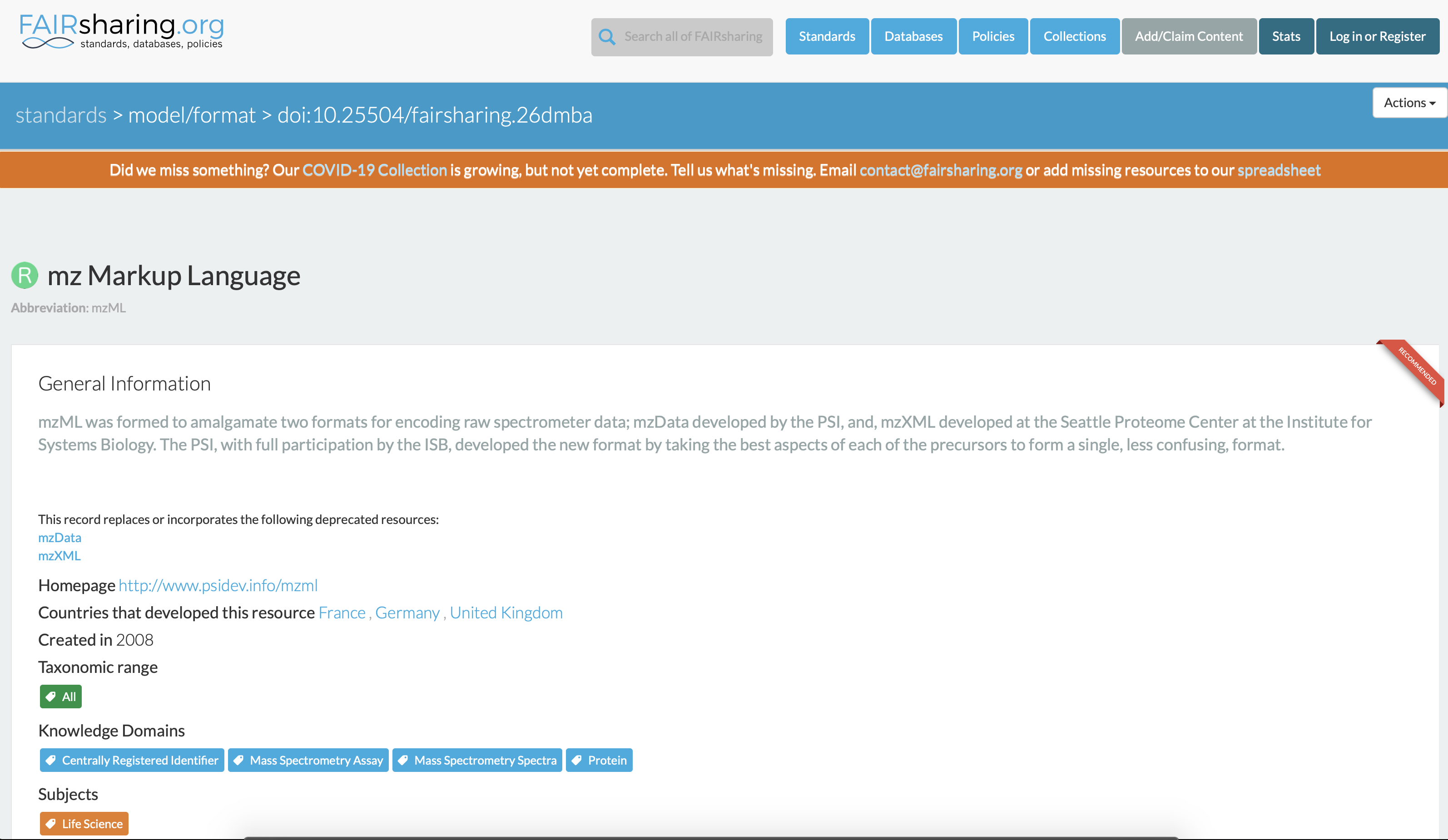
Fig. 10.2 The HUPI-PSI mzML Standard Record in the Elixir FAIRsharing catalog of resources.¶
The objective here is to conversion raw data in manufacturer format to an open format, which would allow data to be used without restrictions. To achieve this, we rely on a containerized version of the Proteowizard 2.
requirements:
10.7.3.1. 1. install docker:¶
on a MacOS system, invoke the following:
>brew update
>brew install docker
10.7.3.2. 2. start and sign in to docker¶
>docker start
>docker login
10.7.3.3. 3. sign-up and/or login to https://hub.docker.com/¶
10.7.3.4. 4. pull the docker container for ProteoWizard¶
:warning: be sure to sign-up and login to https://hub.docker.com/
>docker pull chambm/pwiz-i-agree-to-the-vendor-licenses
In order to be able to reach
https://hub.docker.com/r/chambm/pwiz-skyline-i-agree-to-the-vendor-licenses
Run the Proteowizard
pwiz toolfrom the container over the WATERS raw data, by issueing the following command from the terminal:
>docker run -it --rm -e WINEDEBUG=-all -v /Users/bob/Documents/IMI-FAIR+/Resolute/RESOLUTE_Targted_Metabolomics_of_parental_cell_lines/Raw/MCC025_p180_102024_102059_20190125/raw\ data/KIT2-0-8015_1024503073/:/data chambm/pwiz-skyline-i-agree-to-the-vendor-licenses wine msconvert /data/KIT2-0-8015_1024503073_01_0_1_1_10_11000002.raw --mzML
The command explained:
By essence, the resulting mzML files generated during the conversion are syntactically valid documents as the pwiz performs validation against the mzml xml schema during the serialization.
In some situations, the conversion will fails and no mzML output will be generated. Various reasons can explain failure to convert. The most common ones are corrupted:
raw data files
unsupported vendor format
To address the former, it is good practice to compute a hash (md5, sha2) checksum fingerprinting each of the files. This allows to ensure that no file corruption has occurred during transfer and copy.
To address the latter, one should consult the table of compatibility:
Format |
Status |
|---|---|
ABI T2D |
not working |
Agilent MHDAC (non-IMS) |
working |
Agilent MIDAC (IMS) |
working |
Bruker BAF |
working |
Bruker FID/YEP |
not working |
Bruker TDF |
working |
Sciex WIFF |
working |
Sciex WIFF2 |
working |
Shimadzu QQQ |
working |
Shimadzu QTOF |
working |
Thermo RAW |
working |
Waters RAW |
working |
Waters UNIFI |
not working |
10.7.3.5. 5. Testing and processing the resulting mzML files¶
For users unfamiliar with format, a search via popular search engine will yield options. Alternately, users may consult the Elixir Biotools registry for suggestions.
A number of libraries are available for parsing (reading and writing) mzML document. mzML is a king of XML format for which an XML schema has been defined and allows syntactic validation through standard library in languages such as java, c++ or python. The top hit corresponds the the pymzml library 1.
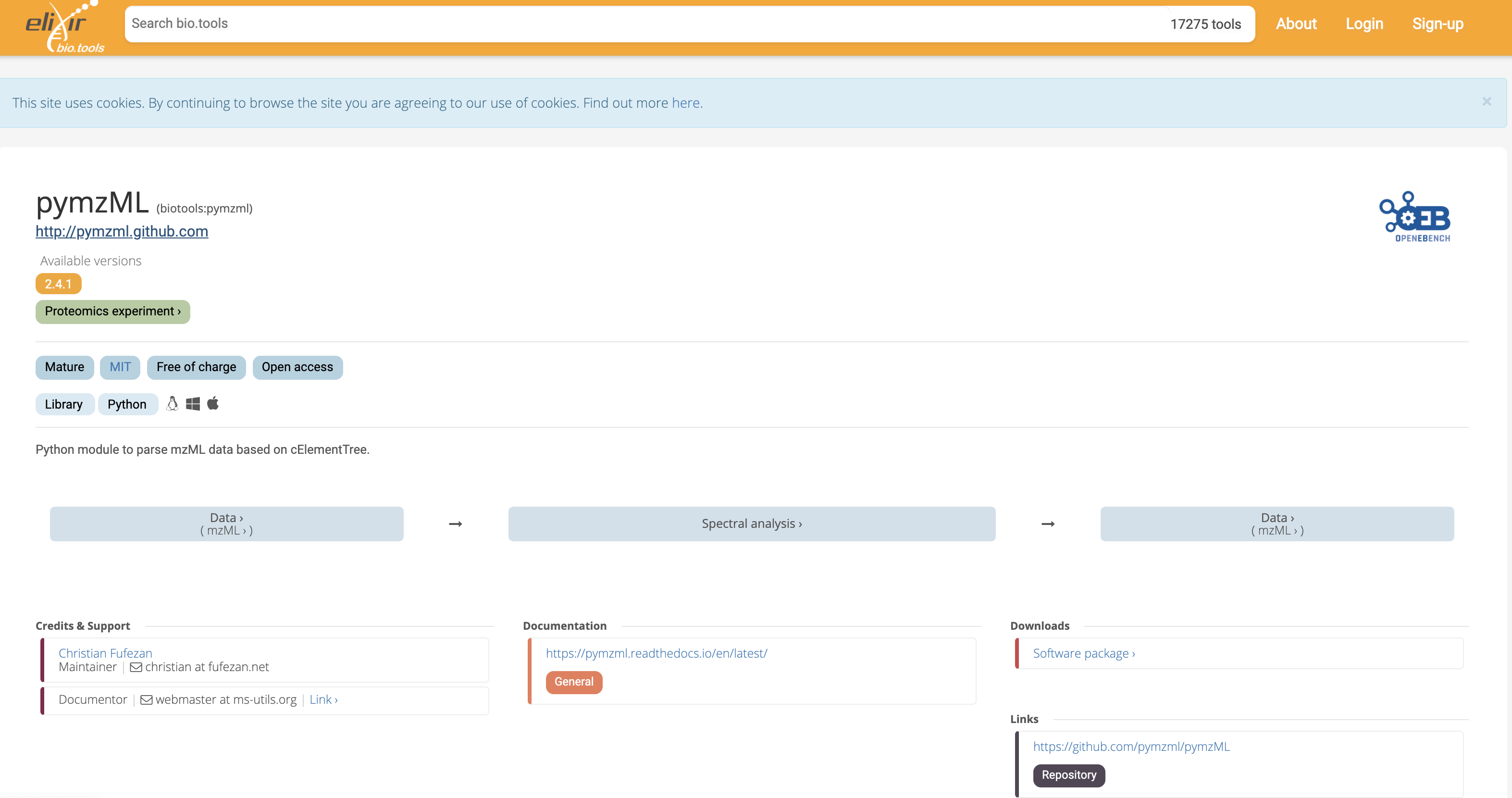
Fig. 10.3 The Python pymzml library entry in the Elixir Biotools catalog of resources.¶
import os
import sys
import pymzml
from pymzml.plot import Factory
mzml_file ="KIT3-0-8015_1024503088_42_0_1_1_00_1024502932.mzML"
msrun = pymzml.run.Reader(mzml_file)
mzml_basename = os.path.basename(mzml_file)
pf = Factory()
pf.new_plot()
pf.add(msrun["TIC"].peaks(), color=(0, 0, 0), style="lines", title=mzml_basename)
pf.save(
"chromatogram_{0}.html".format(mzml_basename),
layout={"xaxis": {"title": "Retention time"}, "yaxis": {"title": "TIC"}},
)
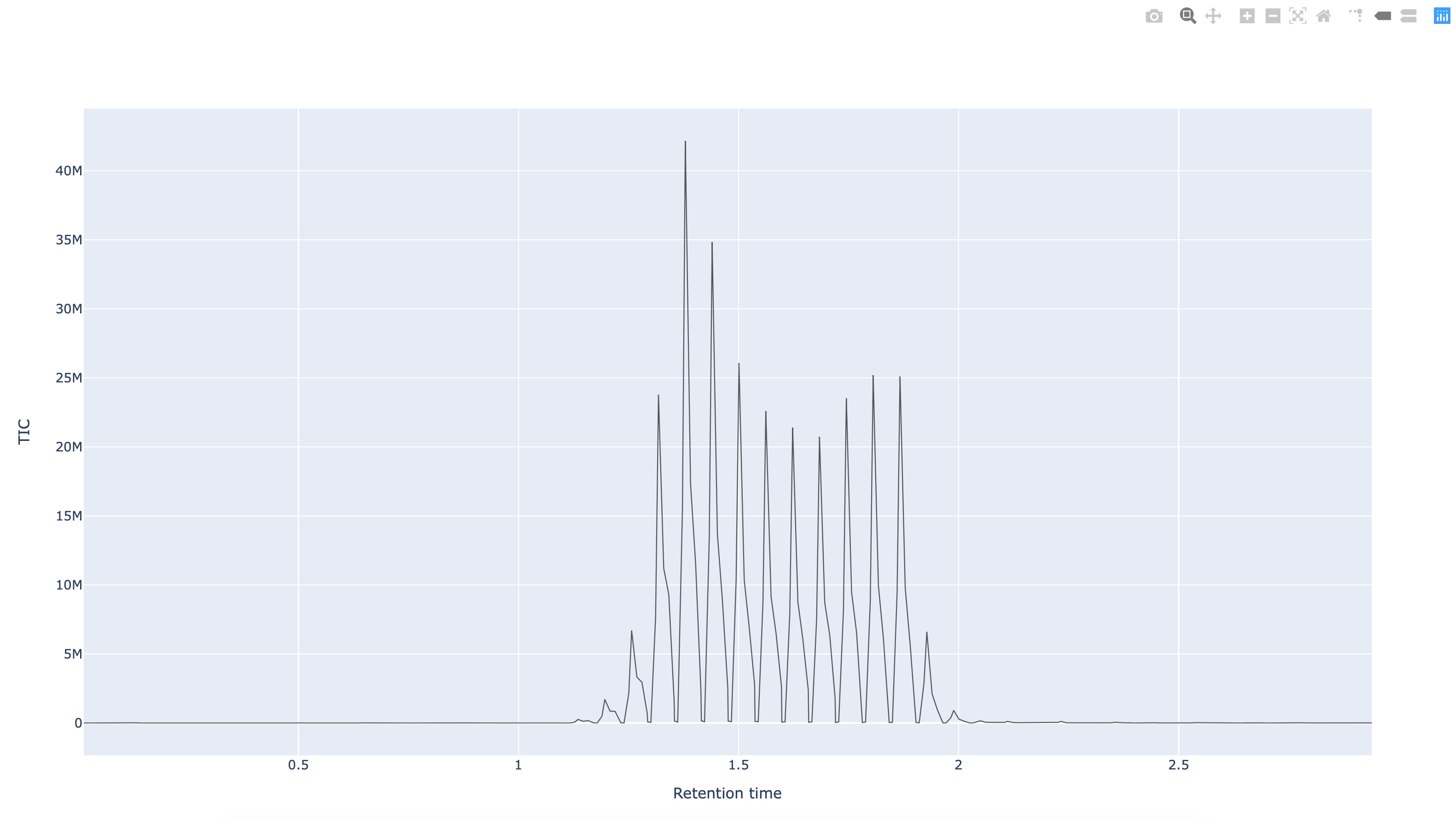
Fig. 10.4 The Python pymzml library rendering of a spectrum as extracted from an mzml formatted mass spectrum data file.¶
In the follow-up recipe, we will show how to boostrap the creation of an ISA metadata file from a bag of mzML documents. But in the following, we’ll show how to read an mzML file using a python library.
10.8. Conclusion¶
In this recipe, we have shown how to convert a proprietary file format to an open standard format, using the exemplar situation of mass spectrometry data. Of course, there are many domain specific data formats and unfortunately not all benefit from the support of open source / open format communities. However by consulting the Elixir UK FAIRsharing registry, it is possible to identify if such open format specifications are available. Then, interrogating the Biotools catalog, it may well be also possible to retrieve libraries and software components allowing manipulations of such format.
10.8.1. What to read next¶
How to package data for shipping with BDbags
How to produce an ISA metadata file from a set of mzML fles
10.9. References¶
- 1
T. Bald, J. Barth, A. Niehues, M. Specht, M. Hippler, and C. Fufezan. pymzML–Python module for high-throughput bioinformatics on mass spectrometry data. Bioinformatics, 28(7):1052–1053, Apr 2012.
- 2
M. C. Chambers, B. Maclean, R. Burke, D. Amodei, D. L. Ruderman, S. Neumann, L. Gatto, B. Fischer, B. Pratt, J. Egertson, K. Hoff, D. Kessner, N. Tasman, N. Shulman, B. Frewen, T. A. Baker, M. Y. Brusniak, C. Paulse, D. Creasy, L. Flashner, K. Kani, C. Moulding, S. L. Seymour, L. M. Nuwaysir, B. Lefebvre, F. Kuhlmann, J. Roark, P. Rainer, S. Detlev, T. Hemenway, A. Huhmer, J. Langridge, B. Connolly, T. Chadick, K. Holly, J. Eckels, E. W. Deutsch, R. L. Moritz, J. E. Katz, D. B. Agus, M. MacCoss, D. L. Tabb, and P. Mallick. A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol, 30(10):918–920, Oct 2012.
10.10. Authors¶
Authors
Name |
ORCID |
Affiliation |
Type |
ELIXIR Node |
Contribution |
|---|---|---|---|---|---|
University of Oxford |
|
Writing - Original Draft |
|||
University of Oxford |
|
Writing - Review & Editing, Funding acquisition |
|||
Bayer AG |
Writing - Review & Editing |