14. Converting VCF file to HL7 FHIR JSON Genomic Report profile¶

14.1. Main Objectives¶
The main purpose of this recipe is to provide FAIR guidance relevant to the clinical domain by:
providing a tool to convert Variant Call Files (VCF) to a HL7 FHIR message
highlighting known limitations of the solution
raising awareness of the FHIR standard in the context of clinically relevant data.
discussing the benefits of obtaining genetic variation information in a regularized form and available in a well-known syntax.
14.2. Graphical Overview¶
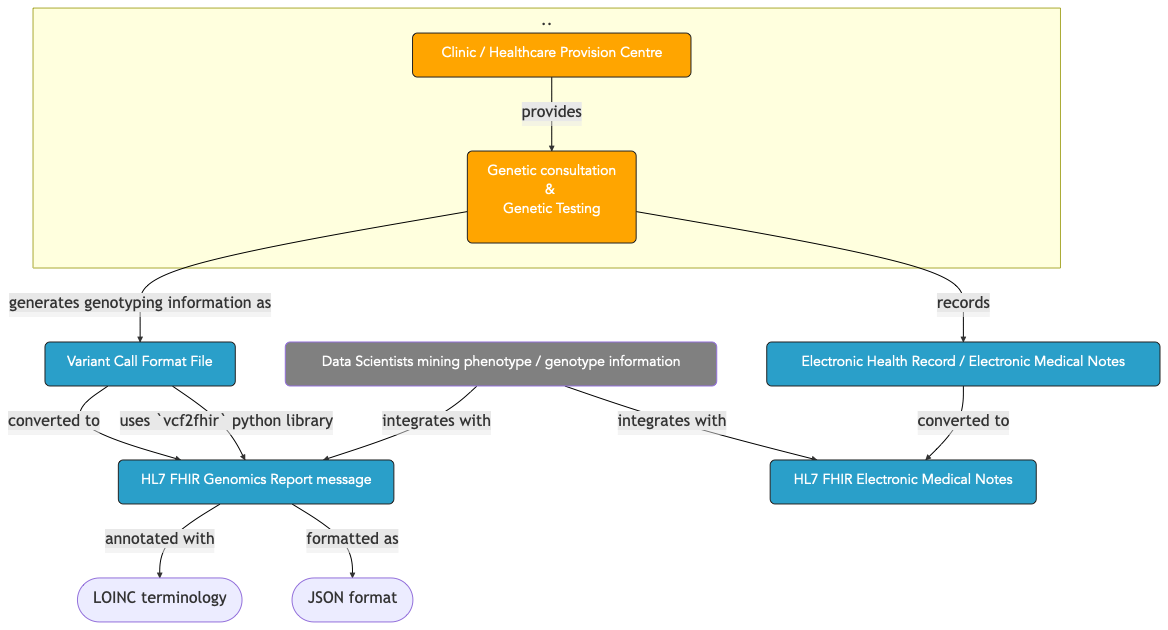
Fig. 14.1 Context for a scenario requiring converting a VCF open standard file to a HL7 FHIR formatted payload.¶
14.3. User Stories¶
The table below lists relevant use cases.
As a .. |
I want to .. |
So that I can .. |
|---|---|---|
Data owner |
Convert VCF to a FHIR message |
Produce an information payload carrying patient genotyping information in a standardized format compatible with EHR |
Data consumer |
Integrate patient genetic information |
Have seamless integration with other FHIR messages from other sources |
Data manager |
Unify clinical information in one format |
Facilitate reuse and mining by clinicians |
14.4. Capability & Maturity Table¶
Capability |
Initial Maturity Level |
Final Maturity Level |
|---|---|---|
Interoperability |
minimal |
repeatable |
14.5. FAIRification Objectives, Inputs and Outputs¶
Actions.Objectives.Tasks |
Input |
Output |
|---|---|---|
Conversion results, Error report |
14.6. Table of Data Standards¶
Data Formats |
Terminologies |
Models |
|---|---|---|
The Variant Call File or VCF is a file format specified by the Global Alliance for Genomic Health to report on genetic variation as detected by a range of molecular biology techniques (e.g., PCR, GeneChip, nucleic acid sequencing).
It is considered to be the de facto standard for reporting genetic variations in their various forms. It is therefore the output for most genetic analysis pipelines (e.g., the Galaxy Worflow tool affy2vcf )
The latest version of Variant Call File format is v4.3, the detailed specifications of which can be found here
The VCF format is species agnostic, making it suitable for use in any context, from agronomy to clinical practice. In fact, it is this last use case that this particular recipe will be focusing on. Indeed, this is when bioinformatics meets medical informatics and the need to translate data into different format arises. In the world of clinical informatics, exchanging information between systems increasingly relies on Health Level 7 data standards and in particular on the Fast Healthcare Interoperability Resource (FHIR). A number of working groups focus on how to best fit clinical information within the paradigm of the HL7 FHIR representation.
In this FAIR Cookbook recipe, we will highlight a software component allowing to convert a specific type of genetic variation information stored in VCF files into an HL7 FHIR compliant, JSON formated message.
The aptly named vcf2fhir library is a python package designed to perform this task. It is the result of work recently published by Dolin et al, 2021. 1.
Warning
In its current form, vcf2fhir :
supports
simple variants(SNVs, MNVs, Indels)does not support
structural variantsThis software is not intended for use in production systems
This software is mainly a prototype for evaluation, testing and refinement of the conversion process and the FHIR message payload
This software does not deal with patient reidentification issues, which needs to be carefully considered if dealing with real patient data, and not test data.
14.6.1. Requirements¶
Users should be at ease with command line interfaces.
In order for the vcf2fhir python library to run, the following libraries need to be present on the system.
Software |
Description |
Version |
|---|---|---|
C-Extensions for Python |
0.29.24 |
|
wheel |
0.37.0 |
|
File downloader |
1.19.4 |
Users running a python environment (virtual or otherwise) need to install cython and wheel, which respectively provide C-implemented extensions for speed and a code packaging library which allows installations to run without the need for building and compiling. The ‘wget’ component will be used to obtain test files from a web address via a command line call.
Both python modules can be installed with a single invokation of the python installation pip command as follows:
pip install cython wheel
14.6.2. Installing vcf2fhir python library:¶
Since the vcf2fhir is available from pypi.org, we can also install the vcf2fhir binary using pip.
pip install vcf2fhir
14.6.3. Using vcf2fhir package:¶
In order to use the VCF to FHIR converter function provided by the library, one needs to first obtain VCF files.
Not only that, but as we indicated in the introduction, the version of the VCF files should be at least v4.1. Furthermore, they should be such that they contain only simple variant information and not structural variants (a type of genetic variations which aren’t currently supported by the vcf2fhir library).
Note
The following section shows how to download VCF files available from public location, the VCF2FHIR github repository in this instance.
Obtaining an exemplar VCF file from the vcf2fhir github repository using the wget command:
wget -c https://raw.githubusercontent.com/elimuinformatics/vcf2fhir/master/vcf2fhir/test/vcf_example1.vcf
more vcf_example1
will show the following:
##fileformat=VCFv4.1
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT TEST001
1 15 . . G . . . GT:PS 0/1:.
1 16 . A,T G . . . GT:PS 0/1:.
1 17 . A G . 27 . GT:PS 0/1:.
1 18 . A G . . SVTYPE=INV GT:PS 0/1:.
1 20 . G . . . GT:PS 0/1:.
1 25 . A G . . . PS .
1 26 . A G . . . GT:PS ./.:.
1 27 . A G . . . GT:PS .|.:.
1 28 . A <CGA_CNVWIN> . . . GT:PS 0/1:.
chr1 301 . A G . . . GT:PS 0/1:.
1 400 . A G . . . GT:PS 1/1:.
1 500 . A G . PASS . GT:PS 0|1:500
1 600 . A G . . . GT:PS 1|0:500
1 700 . A G . . . GT:PS 0|1:700
14.6.4. Conversion from VCF to FHIR¶
The conversion command can be issued as follows:
import vcf2fhir
vcf_fhir_converter = vcf2fhir.Converter('vcf_example1.vcf', 'GRCh37')
vcf_fhir_converter.convert()
The result of the conversion is a so-called FHIR Genomics report, the specifications of which are available here. A number of options are available from the converter to allow users to modify and tune the output to contains specific information depending on the use cases. The conversion can be restricted to a subset of records found in a VCF file by specifying particular portions, e.g., conversion regions, studied regions, clinical annotations, or uncallable regions.
For a full and detailed overview of these options, we direct the readers to the original manual for the vcf2fhir library.
Depending on the options specified by the user, different types of ‘FHIR genomics report’ may be generated. They will differ in content and layout but all rely on a number of normative patterns and terminologies (e.g. LOINC).
Create FHIR Diagnostic Report
Create RegionStudied observations
Create Variant observations
Create SequencePhaseRelationship observations
Create DiagnosticImplication observations
More examples to instantiate the converter
Converts all variants in VCF. FHIR report contains no region-studied observation.
vcf2fhir.Converter('vcftests.vcf','GRCh37', 'aabc')
Converts all variants in VCF. FHIR report assign homoplasmic vs. heteroplasmic based on:
If allelic depth (FORMAT.AD)/ read depth (FORMAT.DP) is greater than 0.89 then allelic state is homoplasmic; otherwise, it is heteroplasmic.
Note : the default value of ratio_ad_dp = 0.99 and the ratio_ad_dp is considered valid only when its value lies between 0 and 1.
vcf2fhir.Converter('vcftests.vcf','GRCh37', 'aabc', ratio_ad_dp = 0.89)
Submitting only noncallable region without other regions generates an error.
vcf2fhir.Converter('vcftests.vcf','GRCh37', 'babc', nocall_filename='WGS_b37_region_noncallable.bed')
Converts all variants in VCF. FHIR report contains one region-studied observation per studied chromosome.
vcf2fhir.Converter('vcftests.vcf','GRCh37', 'cabc', region_studied_filename='WGS_b37_region_studied.bed')
Converts all variants in VCF. FHIR report contains one region-studied observation per studied chromosome.
vcf2fhir.Converter('vcftests.vcf','GRCh37', 'dabc', region_studied_filename='WGS_b37_region_studied.bed', nocall_filename='WGS_b37_region_noncallable.bed')
Converts all variants in conversion region. FHIR report contains no region-studied observation.
vcf2fhir.Converter('vcftests.vcf','GRCh37', 'eabc', conv_region_filename='WGS_b37_convert_everything.bed')
Submitting only noncallable region without other regions generates an error.
vcf2fhir.Converter('vcftests.vcf','GRCh37', 'fabc', conv_region_filename='WGS_b37_convert_everything.bed', nocall_filename='WGS_b37_region_noncallable.bed')
Converts all variants in conversion region. FHIR report contains one region-studied observation per studied chromosome, intersected with conversion region.
vcf2fhir.Converter('vcftests.vcf','GRCh37', 'gabc', conv_region_filename='WGS_b37_convert_everything.bed', region_studied_filename='WGS_b37_region_studied.bed')
Converts all variants in conversion region. FHIR report contains one region-studied observation per studied chromosome, intersected with conversion region.
vcf2fhir.Converter('vcftests.vcf','GRCh37', 'habc', conv_region_filename='WGS_b37_convert_everything.bed', region_studied_filename='WGS_b37_region_studied.bed', nocall_filename='WGS_b37_region_noncallable.bed')
Conversion of a bgzipped VCF
vcf2fhir.Converter('vcf_example4.vcf.gz','GRCh37', 'kabc', has_tabix=True)
Below is a typical output of the vcf2fhir tool: a HL7 FHIR message compliant with the Genomics Report pattern.
Note the use of LOINC terminology for key descriptors.
{
"resourceType": "DiagnosticReport",
"id": "",
"meta": {
"profile": [
"http://hl7.org/fhir/uv/genomics-reporting/StructureDefinition/genomics-report"
]
},
"contained": [
{
"resourceType": "Observation",
"id": "",
"meta": {
"profile": [
"http://hl7.org/fhir/uv/genomics-reporting/StructureDefinition/region-studied"
]
},
"status": "final",
"category": [
{
"coding": [
{
"system": "http://terminology.hl7.org/CodeSystem/observation-category",
"code": "laboratory"
}
]
}
],
"code": {
"coding": [
{
"system": "http://loinc.org",
"code": "53041-0",
"display": "DNA region of interest panel"
}
]
},
"subject": {
"reference": "Patient/NB6TK328"
},
"component": [
{
"code": {
"coding": [
{
"system": "http://loinc.org",
"code": "92822-6",
"display": "Genomic coord system"
}
]
},
"valueCodeableConcept": {
"coding": [
{
"system": "http://loinc.org",
"code": "LA30102-0",
"display": "1-based character counting"
}
]
}
},
{
"code": {
"coding": [
{
"system": "http://loinc.org",
"code": "48013-7",
"display": "Genomic reference sequence ID"
}
]
},
"valueCodeableConcept": {
"coding": [
{
"system": "http://www.ncbi.nlm.nih.gov/nuccore",
"code": "NC_000001.11"
}
]
}
},
{
"code": {
"coding": [
{
"system": "http://loinc.org",
"code": "51959-5",
"display": "Range(s) of DNA sequence examined"
}
]
},
"valueRange": {
"high": {
"value": 201113567.0
},
"low": {
"value": 201038512.0
}
}
},
{
"code": {
"coding": [
{
"system": "http://loinc.org",
"code": "51959-5",
"display": "Range(s) of DNA sequence examined"
}
]
},
"valueRange": {
"high": {
"value": 201378701.0
},
"low": {
"value": 201358008.0
}
}
}
]
}
]
}
14.6.4.1. Built-in Support Conversion Error Logging¶
No conversion tool is failsafe. Therefore, the vcf2fhir library provides 2 distinct logging functions, which plug naturally into the python generic error logging package.
vcf2fhir.general: this mode provides the standard library logging functions.
vcf2fhir.invalidrecord: this mode logs all the
recordsfrom the input vcf file which are in present in theconversion regionbut are not converted tofhir format.
To take advantage of this mechanism, users can invoke each of the vcf2fhir loggers in the manner described below:
import logging
# create logger
logger = logging.getLogger('vcf2fhir.invalidrecord')
logger.setLevel(logging.DEBUG)
# create console handler and set level to debug
ch = logging.FileHandler('invalidrecord.log')
ch.setLevel(logging.DEBUG)
# create formatter
formatter = logging.Formatter('%(asctime)s - %(name)s - %(levelname)s - %(message)s')
# add formatter to ch
ch.setFormatter(formatter)
# add ch to logger
logger.addHandler(ch)
14.6.5. Take home message from using the vcf2fhir python library¶
an
initial capabilitysupporting generation ofHL7 FHIR Genomics Report messagefrom VCF files.generation of LOINC-annotated, JSON formatted documents containing simple genetic variation information.
availability of a conversion error log, for quality control and error tracking tasks.
14.7. Conclusion¶
Why does this matter and how does it relate to FAIR?
The conversion from VCF to HL7 FHIR JSON message has to do with the **I and R** of FAIR, that is interoperability and reusability.
From a syntactic standpoint, the availability of genetic variation information at a granular level in an easily parseable form (JSON) is a gain for anyone looking at merging this information with other clinical messages.
From a semantic standpoint, the reliance on
LOINCvocabulary to mark up the patterns defined in the HL7 FHIR Genomics Reports enhances interoperation between systems by provided unambiguous annotations.Finally, as more systems are able to produce FHIR messages from a variety of instruments or data sources, the availability of a FHIR message covering a subset of genetic variation available from testing facilities makes investigating and mining phenotypic and genotypic relations more straightforward.
However, one needs to remember that the capability afforded by the
vcf2fhirlibrary is at an early stage and only supports simple cases. More efforts and more efforts is needed before a functionality is available at a Technical Readiness Level compatible with production systems.
Any other important issues?
We have highlighted the existing limitations surrounding the use of the open source conversion tool and that users should carefully assess the nature of the information present in the input VCF files prior to executing the code. Bearing this in mind, the
vcf2fhirtool provides an easy to deploy and easy to use solution for anyone interested in adding a FHIR message capability to a clinical genetic analysis pipeline,for instance on consuming DNA microarray GeneChip genotyping solutions. The authors of the tool aim to expand its capabilities to includeenhancing the conversion logic to accommodate VCF rows representing structural variants (i.e. rows that contain an INFO.SVTYPE field).Finally, it is important to realize that the resulting JSON message, as it is, lacks important metadata to be fully and properly FAIR (e.g.,
licence information). One has therefore to see thiscapabilityas one of the many elements that needs to be put together to build and deliver a FAIR infrastructure. For instance, this HL7 JSON message could be embedded in a more complex system, which would package information and deliver a FAIR payload.
14.7.1. What to read next¶
Pistoia Alliance FAIR4Clinical Guidance - An Introduction
Pistoia Alliance FAIR4Clin - Metadata
14.8. References¶
References
- 1
R. H. Dolin, S. R. Gothi, A. Boxwala, B. S. E. Heale, A. Husami, J. Jones, H. Khangar, S. Londhe, F. Naeymi-Rad, S. Rao, B. Rapchak, J. Shalaby, V. Suraj, N. Xie, S. Chamala, and G. Alterovitz. vcf2fhir: a utility to convert VCF files into HL7 FHIR format for genomics-EHR integration. BMC Bioinformatics, 22(1):104, Mar 2021.
14.9. Authors¶
Authors
Name |
ORCID |
Affiliation |
Type |
ELIXIR Node |
Contribution |
|---|---|---|---|---|---|
University of Oxford |
|
Writing - Original Draft |
|||
SIB |
|
Writing - Review & Editing |
|||
University of Luxembourg |
|
Writing - Review & Editing |