12. File format validation, FASTQ example¶

12.1. Main Objectives¶
The main purpose of this recipe is to:
provide a FASTQ file validation solution
propose a general file validation workflow.
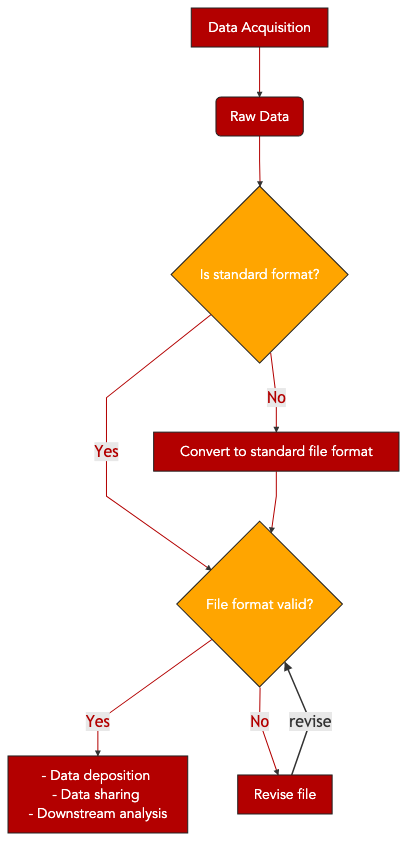
12.3. User Stories¶
The table below lists common file validation use cases. This recipe provides solutions with FASTQ files 1 as an example.
As a .. |
I want to .. |
So that I can .. |
|---|---|---|
Data owner |
Validate my sequencing files before depositing to public archives |
Reduce the risk of submitting invalid files or submission rejection |
Data consumer |
Validate files before running analysis |
Avoid wasting time and resource processing corrupted files |
Data consumer |
Integrate file format validation into my data process pipeline |
Build a more reproducible and error-proof pipeline |
Data librarian |
Check files downloaded from unknown sources before deposition |
Ensure the file is usable in the future. |
12.4. Capability & Maturity Table¶
Capability |
Initial Maturity Level |
Final Maturity Level |
|---|---|---|
Interoperability |
minimal |
repeatable |
12.5. FAIRification Objectives, Inputs and Outputs¶
Actions.Objectives.Tasks |
Input |
Output |
|---|---|---|
Validation results |
12.6. Table of Data Standards¶
Data Formats |
Terminologies |
Models |
|---|---|---|
FASTQ is the de facto sequencing file format and one of the most common file formats in bioinformatics analysis 2, 4. Researchers receive FASTQ files from various sources. These files are used intensively in automated bioinformatics analysis pipelines. Therefore, it is important to validate FASTQ files to improve the data reusability and build error-proof data analysis processes.
FASTQ validators detect truncated reads, base calls and quality score mismatches, invalid encoding, etc. For paired-end reads, they also check if the forward reads match with the reverse reads. Most validators can process different FASTQ variants automatically and handle compressed FASTQ files.
FASTQ-utils is an open-source software to validate and process FASTQ files. It has been applied in the European Nucleotide Archive(ENA), and several research initiatives.
This recipe provides an example of validating FASTQ files with FASTQ-utils on MacOS and Linux machines.
Warning
⚠️ Quality control is out of the scope of file format validation.
Fig. 12.2 the FASTQutils library.¶
12.6.1. Requirements¶
The users are expected to be comfortable with Unix-based OS and basic Bash programming syntax and commands.
Software |
Description |
Version |
|---|---|---|
Package manager for installing validators |
4.8.3 |
|
FASTQ validator |
0.23.0 |
|
File downloader |
1.19.4 |
12.6.2. Step 1: Install fastq-utils¶
The command below installs fastq-utils via Conda. It is also possible to install fastq-utils from the source code 3.
conda install -c bioconda fastq_utils
12.6.3. Step 2: Get example file for testing*¶
Note
Users can skip this step and test with their own files._
In this step, we download example FASTQ files from ENA for testing. The first example file is a single read file, the other ones are paired-end read files.
Example 1: Get single read FASTQ file
The command below downloads an Ion Torrent S5 fastq file from ENA. This file is the whole genome sequencing file of SARS-CoV-2. The complete file is 192Mb.
wget -c ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR121/077/SRR12132977/SRR12132977.fastq.gz
Users can inspect the fastq.gz file using gzip -cd SRR12132977.fastq.gz | head -8. Below is the header of this FASTQ file.
@SRR12132977.1 1/1
AACAAACCAACCAACTTTCGATCTCTTGTAGATCTGTTCTCTAAACGAACAAACTAAAATGTCTGATAATGGACCCCAAAATCAGCGAAATGCACCCCGCATTACGTTTGGTGGACCCTCAG
+
C@CCD>DBC?B692;;;09?<BBBBC>BBBBBBBBB@?ABB@BC<BBB>@A?:999992;=>>@??==:=C;>=<:'555)8;;;;;AG:AAAAADD;CCBB>?@;;;0:<@A>CEE?CFCC
@SRR12132977.2 2/1
AACAAACCAACCAACTTTCGATCTCTTGTAGATCTGTTCTCTAAACGAACTTTAAAATCTGTGTGGCTGTCACTCGGCTGCATGCTTAGTGCACTCACGCAGTATAATTAATAACTAATTACTGTCGTTGACAGGACACGAGTAACTCGTCTATCTTCTGCAGGCTGCTTACGGTTTCGTCCGTGTTGCAGCCGATCATCAGCAC
+
A>A@@=@@F@D@C<999,:<@ABBBB@B=>=BB@BBB?@@><;;7>??=BBB>BDD;D>????@@;@CDC@@@BBB>BBB@AAC>>9BBBB;;;@@?;><::;99<9<;A;>><@@A:=:>@@@>A@>:>===>:=<<>>;;;>=BCAA?>=A>>>:==>;998<=;===@@@<>>9>>>?;??==:=>>>>:>>;;;;;;;<;;
Example 2: Get paired-read FASTQ files
The command below downloads Illumina iSeq 100 paired end sequencing files from ENA. These files are raw sequence reads of a SARS-CoV-2 sample. Each file is 26 Mb.
wget -c \
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR115/044/SRR11542244/SRR11542244_1.fastq.gz \
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR115/044/SRR11542244/SRR11542244_2.fastq.gz
Below is the headers of the two files. The read pairs info is listed in the read IDs.
# Header of the forward read, SRR11542244_1.fastq.gz
@SRR11542244.1 1/1
GTGTGTGTATACATATATATATATATCACATTTTCTTTATCCATTTATCTGTTGTTGGACACTTAGGTTGATTCCATATCTTGGCTATTGTGAATAGTG
+
,,FFFFFFFFFFFFFFFFFFFFFFFF:FFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFF
@SRR11542244.2 2/1
GTGATTCCTCAAAGATTTAGAACCAGAAATACCATGTGACCCAGCAATTCCATTACCAGGTCTAAACCCAAAGGAATATAAATCATTCTGTAATGAAGATA
+
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
# Header of the reverse read, SRR11542244_2.fastq.gz
@SRR11542244.1 1/2
CTATTGGGTATTTAATCCAAAGAAAGGAAATCGGTATATCAAAGAGACATCTGCATGCCCATGTTTATTGTAGCACTATTCACAATAGCCAAGATATGGAA
+
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFF
@SRR11542244.2 2/2
GAACATATGTGTGCATGTATCTTCATTACAGAATGATTTATATTCCTTTGGGTTTAGACCTGGTAATGGAATTGCTGGGTCACATGGTATTTCTGGTTCTA
+
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
12.6.4. Step 3: Perform validation¶
The command below validates the single read file in Example 1.
fastq_info -r SRR12132977.fastq.gz
Below are the validation results. fastq-utils returns the number of reads, read length details, and encoding info. Field Quality encoding indicates the fastq file variant. FASTQ-utils returns OK for a valid fastq file. Otherwise, it will return the validation details in the Error message.
Skipping check for duplicated read names
1900000
------------------------------------
Number of reads: 1919741
Quality encoding range: 34 77
Quality encoding: 33
Read length: 25 352 215
OK
The validation of paired end reads is similar to single read file validation.
fastq_info SRR11542244_1.fastq.gz SRR11542244_2.fastq.gz
Here are the validation results.
fastq_utils 0.23.0
DEFAULT_HASHSIZE=39000001
Scanning and indexing all reads from SRR11542244_1.fastq.gz
700000Scanning complete.
Reads processed: 733611
Memory used in indexing: ~47 MB
File SRR11542244_1.fastq.gz processed
Next file SRR11542244_2.fastq.gz
700000
------------------------------------
Number of reads: 733611
Quality encoding range: 35 70
Quality encoding: 33
Read length: 35 101 96
OK
fastq_util also provides additional arguments to tune the validation:
-s: to validate if reads in two files have the same ordering.
-r: to skip duplicated read names validation. It uses less memory and runs faster.
-e: to allow empty files pass the validation
-q: not to fail if the encoding can’t be decided.
12.6.4.1. Error messages for invalid files¶
FASTQ-utils returns an error message with the location of invalid lines and type of errors if the files are invalid. Below are examples of error messages.
Invalid file example 1, duplicated reads
ERROR: Error in file SRR11542244_2.fastq: line 16: duplicated sequence SRR11542244.5 5/
Invalid file example 2, wrong base call encoding
ERROR: Error in file SRR11542244_2.fastq: line 5: invalid character ‘e’ (hex. code:’65’), expected ACGTacgt0123nN.
12.6.5. FASTA-utils feature summary¶
The table lists technical considerations when selecting the validator, including basic validation function, performance, interface, etc. It also provides a detailed summary of fastq-utils features.
Aspects |
Validation content |
Description |
FASTQ-utils |
|---|---|---|---|
Basic validation |
4-line format |
Check if the FASTQ file is a 4-line file |
☑️ |
Character encoding |
Check if the base calls and quality score encoding are correct. |
☑️ |
|
Read length |
Check if the length of the base calls are the same as that of the quality scores |
☑️ |
|
File truncation |
Check if the file is truncated or not |
☑️ |
|
Paired-end reads validation |
Deinterleaved paired reads |
Validate when the forward and reverse reads are in two files. |
☑️ |
Interleaved “8-line” files |
Validate when the forward and reverse reads are listed together as an 8-line file |
☑️ |
|
Compressed file validation |
gzip |
Validate compressed fastq files, with extension |
☑️ |
FASTQ variants* validation |
fastq-illumina |
Validate the fastq-illumina format |
☑️ |
fastq-sanger |
Validate the fastq-sanger format |
☑️ |
|
fastq-solexa |
Validate the fastq-solexa format |
☑️ |
|
Performance |
Memory |
|
|
Speed |
|
||
Archieve compatiablity |
ENA |
File validated can be submitted to the ENA archive. |
☑️ |
ArrayExpress |
File validated can be submitted to Array Express. |
☑️ |
|
SRA |
File validated can be submitted to the SRA archive. |
☑️ |
|
Interface |
Command line interface |
Can be used in shell and intergerated in pipe commands |
☑️ |
License |
Licensed |
☑️GPL-3 |
|
Commercial use |
Can be used for commercial purpose |
☑️ |
|
Code |
Open source |
Source code available on public platforms |
☑️ |
*See details in the [FASTQ specification recipe]( TODO include link).
12.7. Conclusion¶
In this recipe, we have shown how to validate fastq files, and proposed indicators to evaluate a FASTQ validator. We also identified common file validation related use cases and provided a general file validation workflow. This recipe can be expanded to other file formats and other use cases.
12.7.1. What to read next¶
🐙From proprietary format to open standard format: an exemplar
🐙[FASTQ file specification recipe](TODO include link to recipe https://www.TBD.org )
🐙[FASTQ file validator in Biopython](TODO include link to recipe https://www.TBD.org)
12.8. References¶
References
- 1
P. J. Cock, C. J. Fields, N. Goto, M. L. Heuer, and P. M. Rice. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res, 38(6):1767–1771, Apr 2010.
- 2
ENA. Accepted read data formats — ena training modules 1 documentation. 2020. URL: https://ena-docs.readthedocs.io/en/latest/submit/fileprep/reads.html#fastq-format.
- 3
Nuno Fonseca and Jonathan Manning. Nunofonseca/fastq_utils 0.24.0. jul 2020. URL: https://doi.org/10.5281/zenodo.3936692, doi:10.5281/zenodo.3936692.
- 4
NCBI. File format guide. 2020. URL: https://www.ncbi.nlm.nih.gov/sra/docs/submitformats/#fastq-files.
12.9. Authors¶
Authors
Name |
ORCID |
Affiliation |
Type |
ELIXIR Node |
Contribution |
|---|---|---|---|---|---|
EMBL-EBI |
|
Writing - Original Draft |
|||
Barcelona Supercomputing Centre |
|
Writing - Review & Editing |
|||
GSK |
Writing - Review & Editing |